The Program switch.pl
Introduction
The switch.pl script can be used to design bi-stable structures, i.e.
structures with two almost equally good foldings. For two given structures
there are always a lot of sequences compatible with both structures. If both
structures are reasonably stable you can find sequences where both target
structures have almost equal energy and all other structures have much higher
energies.
Combined with RNAsubopt, barriers and treekin, this is a very
useful tool for designing RNA switches.
The input requires two structures in dot-bracket notation and additionally
you can add a sequence. It is also possible to calculate the switching
function at two different temperatures with option -T and -T2.
Designing a Switch
Now we try to create an RNA switch using switch.pl [Flamm et al., 2001].
First we create our inputfile, then invoke the program using ten optimization
runs (-n 10) and do not allow lonely pairs. Write it out to switch.out
$ cat > switch.in
((((((((......))))))))....((((((((.......))))))))
((((((((((((((((((........)))))))))))))))))).....
$ switch.pl -n 10 --noLP < switch.in > switch.out
switch.out should look similar like this, the first block represents our
bi-stable structures in random order, the second block shows the resulting
sequences ordered by their score.
$ cat switch.out
GGGUGGACGUUUCGGUCCAUCCUUACGGACUGGGGCGUUUACCUAGUCC 0.9656
CAUUUGGCUUGUGUGUCGAAUGGCCCCGGUACGUAGGCUAAAUGUACCG 1.2319
GGGGGGUGCGUUCACACCCCUCAUUUGGUGUGGAUGUGCUUUCUACACU 1.1554
[...]
the resulting sequences are:
CAUUUGGCUUGUGUGUCGAAUGGCCCCGGUACGUAGGCUAAAUGUACCG 1.2319
GGGGGGUGCGUUCACACCCCUCAUUUGGUGUGGAUGUGCUUUCUACACU 1.1554
CGGGUUGUAACUGGAUAGCCUGGAAACUGUUUGGUUGUAAUCCGAACAG 1.0956
[...]
Given all 10 suggestions in our switch.out, we select the one with the
best score with some command line tools to use it as an RNAsubopt input
file and build up the barriers tree.
$ tail -10 switch.out | awk '{print($1)}' | head -n 1 > subopt.in
$ RNAsubopt --noLP -s -e 25 < subopt.in > subopt.out
$ barriers -G RNA-noLP --bsize --rates --minh 2 --max 30 < subopt.out > barriers.out
tail -10 cuts the last 10 lines from the switch.out file and pipes
them into an awk script. The function print($1) echoes only the first
column and this is piped into the head program where the first line, which
equals the best scored sequence, is taken and written into subopt.in.
Then RNAsubopt is called to process our sequence and write the output to
another file which is the input for the barriers calculation.
Below you find an example of the barrier tree calculation above done with the
right settings (connected root) on the left side and the wrong RNAsubobt -e
value on the right. Keep in mind that switch.pl performs a stochastic search
and the output sequences are different every time because there are a lot of
sequences which fit the structure and switch calculates a new one everytime.
Simply try to make sure.

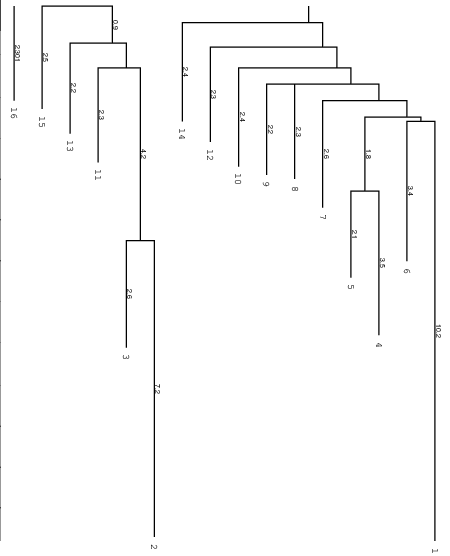
left: Barriers tree as it should look like, all branches connected to the main root
right: disconnected tree due to a too low energy range (-e) parameter set in
RNAsubopt.
Be careful to set the range -e high enough, otherwise we get a problem when
calculation the kinetics using treekin. Every branch should be somehow connected
to the main root of the tree. Try -e 20 and -e 30 to see the difference in
the trees and choose the optimal value. By using --max 30 we shorten our tree
to focus only on the lowest minima.
We then select a branch preferably outside of the two main branches, here branch
30 (may differ from your own calculation). Look at the barrier tree to find the
best branch to start and replace 30 by the branch you would choose. Now use
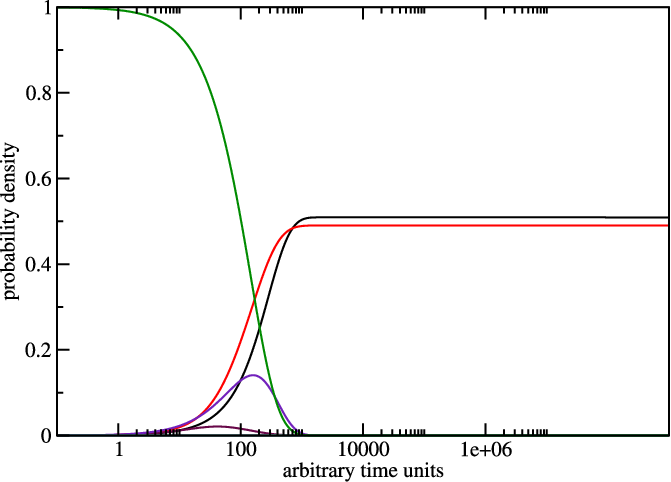
treekin to plot concentration kinetics and think about the graph you just
created.
$ treekin -m I --p0 30=1 < barriers.out > treekin.out
$ xmgrace -log x -nxy treekin.out
The graph could look like the one below, remember everytime you use switch.pl
it can give you different sequences so the output varies too. Here the one from
the example.