
ViennaRNA Package 2 - Documentation
Here, we provide all the documentation necessary to install and use the programs of the ViennaRNA Package. We also provide a reference manual the for RNAlib C library which comes with the package.
Installing the ViennaRNA Package
For best portability the ViennaRNA package uses the GNU autoconf and
automake tools. The instructions below are for installing the ViennaRNA
package from source. However, pre-compiled binaries for various Linux distributions,
as well as for Windows users are available from Download
section of the main ViennaRNA homepage.
Usually you'll just unpack, configure and make. To do this type:
tar -zxvf ViennaRNA-2.7.0.tar.gz cd ViennaRNA-2.7.0 ./configure make sudo make installUser-dir Installation
If you do not have root privileges on your computer, you might want to install
the ViennaRNA Package to a location where you actually have write access to.
To do so, you can set the installation prefix of the ./configure
script like so:
./configure --prefix=/home/username/ViennaRNA make installThis will install the entire ViennaRNA Package into a new directory
ViennaRNA
directly into the users username home directory.
Python interface from PyPI
If you intend to only use the Python bindings to RNAlib you might want
to install them directly from the
Python Package Index
using Pythons pip:
pip install viennarnaNotes for MacOS X users
-
Compilation
Although users will find
/usr/bin/gccand/usr/bin/g++executables in their directory tree, these programs are not at all what they pretend to be. Instead of including the GNU programs, Apple decided to install clang/llvm in disguise. Unfortunately, the default version of clang/llvm does not support OpenMP (yet), but only complains at a late stage of the build process when this support is required. Therefore, it seems necessary to deactivate OpenMP support by passing the option--disable-openmpto the./configurescript. -
Missing EXTERN.h include file
Furthermore, as far as we are informed, users are discouraged to use the Perl 5 interpreter that is shipped with Mac OS X. Instead, one should install a more recent version from another source, e.g.
homebrew. If, however, for any reason you do not want to install your own Perl 5 interpreter but use the one from Apple, you need to specify its include path to enable building the ViennaRNA Perl interface. Otherwise, the fileEXTERN.hwill be missing at compile time. To fix this problem, you first need to find out whereEXTERN.his located:sudo find /Library -type f -name EXTERN.h
Then choose the one that corresponds to your default perl interpreter (find out the version number with
perl -v | grep version), simply execute the following before running the./configurescript, e.g.:export CPATH=/Library/Developer/CommandLineTools/SDKs/MacOSX10.15.sdk/System/Library/Perl/5.18/darwin-thread-multi-2level/CORE
if your default perl is v5.18 running on MacOSX10.15. Change the paths according to your current setup. After that, running
./configureand compilation should run fine. See also these comments on stackoverflow. -
Universal binaries
Additionally, if you intend to build the ViennaRNA such that it runs on both, x86_64 and the armv8 (such as for the M1 processors in recent MacBooks), architectures, you need to build a so-called universal binary. Note, however, that to accomplish this task, you might need to deactivate any third-party library dependency as in most cases, only one architecture will be available at link time. This includes the Perl 5 and Python interfaces but also MPFR and GSL support, possibly even more. In order to compile and link the programs, library, and scripting language interfaces of the ViennaRNA Package for multiple architectures, we've added a configure switch that sets up the required changes automatically:
./configure --enable-universal-binary
Note, that with link time optimization turned on, MacOS X's default compiler (llvm/clang) generates an intermediary binary format that can not easily be combined into a multi-architecture library. Therefore, the
--enable-universal-binaryswitch turns off link time optimization!
Note, that with link time optimization turned on, MacOS X's default compiler (llvm/clang)
generates an intermediary binary format that can not easily be combined into a multi-architecture
library. Therefore, the --enable-universal-binary switch turns off link time
optimization!
Optional sub-packages and configure options
This release includes the RNAforester, Kinfold, and Kinwalker programs,
which can also be obtained as independent packages. Running ./configure
in the ViennaRNA directory will configure these packages as well. However, for
detailed information and compile time options, see the README and
INSTALL files in the respective subdirectories.
Our latest version contains code that implements a faster multibranch loop decomposition in global
MFE predictions, as used e.g. in RNAfold. This implementation makes use of modern processors
capability to execute particular instructions on multiple data simultaneously (SIMD - single instruction
multiple data, thanks to W. B. Langdon for providing the modified code). Consequently, the time required
to assess the minimum of all multibranch loop decompositions is reduced up to about one half compared to
the runtime of the original implementation.
To make use of this piece of code you need
a CPU capable to handle SSE4.1 instructions
and enable the feature at compile-time using the following configure flag:
./configure --enable-sseScripting Interfaces
The ViennaRNA Package comes with scripting language interfaces for Perl 5,
and Python (provided by swig),
that allow one to use the implemented algorithms directly without the need of calling an
executable program. While building the Perl 5 and Python 3 interface is enabled by default,
the interface for Python 2 needs to be explicitely activated. To do so, just pass the
--with-python2 flag to the configure script before running make.
On the other hand, you can build the ViennaRNA package without Perl 5 or Python 3 support
by switching them off at configure time, before the actual installation.
Example:
./configure --without-perl --without-pythonDisabling the entire scripting language support alltogether can be accomplished using the following switch:
./configure --without-swigCluster Analysis
The programs AnalyseSeqs and AnalyseDists offer some
cluster analysis tools (split decomposition, statistical geometry, neighbor joining,
Ward's method) for sequences and distance data. To also build these programs add
--with-cluster to your configure options.
The Kinfold program can be used to simulate the folding dynamics of an
RNA molecule, and is compiled by default. Use the --without-kinfold
option to skip compilation and installation of Kinfold.
The RNAforester program is used for comparing secondary structures using
tree alignment. Similar to Kinfold, use the --without-forester
option to skip compilation and installation of RNAforester.
The Kinwalker algorithm performs co-transcriptional folding of RNAs, starting
at a user specified structure (default: open chain) and ending at the minimum free energy
structure. Compilation and installation of this program is deactivated by default.
Use the --with-kinwalker option to enable building and installation of
Kinwalker.
The RNAlocmin program is part of the Basin Hopping Graph Framework
and reads secondary structures and searches for local minima by performing a gradient walk
from each of those structures. It then outputs an energetically sorted list of local minima
with their energies and number of hits to particular minimum, which corresponds to a size
of a gradient basin. Additional output consists of barrier trees and Arhenius rates to
compute various kinetic aspects.
Compilation and installation of this program is activated by default.
Use the --without-rnalocmin option to disable building and installation
of RNAlocmin.
RNAxplorer is a multitool, that offers different methods to explore RNA energy
landscapes. The main use case is sampling of representative structures of the RNA
conformation space, in order to compute RNA folding kinetics.
Compilation and installation of this program is activated by default.
Use the --without-rnaxplorer option to disable building and installation
of RNAxplorer.
To increase the performance of our implementations, the ViennaRNA Package tries to make use of the Link Time Optimization (LTO) feature of modern C-compilers. If you are experiencing any troubles at make-time or run-time, or the configure script for some reason detects that your compiler supports this feature although it doesn't, you can deactivate it using the flag
./configure --disable-ltoOpenMP support
To enable concurrent computation of our implementations and in some cases parallelization of the algorithms we make use of the OpenMP API. This interface is well understood by most modern compilers. However, in some cases it might be necessary to deactivate OpenMP support and therefore transform RNAlib into a C-library that is not entirely thread-safe. To do so, add the following configure option
./configure --disable-openmpPOSIX threads (pthread) support
To enable concurrent computation of multiple input data in RNAfold, and for our implementation of the concurrent unordered insert, ordered output flush data structure vrna_ostream_t we make use of POSIX threads. This should be supported on all modern platforms and usually does not pose any problems. Unfortunately, we use a threadpool implementation that is not compatible with Microsoft Windows yet. Thus, POSIX thread support can not be activated for Windows builds until we have fixed this problem. If you want to compile RNAfold and RNAlib without POSIX threads support for any other reasons, add the following configure option
./configure --disable-pthreadsSVM Z-score filter in RNALfold
By default, RNALfold that comes with the ViennaRNA Package allows for z-score filtering of its predicted results using a support vector machine (SVM). However, the library we use to implement this feature (libsvm) is statically linked to our own RNAlib. If this introduces any problems for your own third-party programs that link against RNAlib, you can safely switch off the z-scoring implementation using
./configure --without-svmGNU Scientific Library
The new program RNApvmin computes a pseudo-energy pertubation vector
that aims to minimize the discrepancy of predicted, and observed pairing probabilities.
For that purpose it implements several methods to solve the optimization problem.
Many of them are provided by the GNU Scientific Library,
which is why the RNApvmin program, and the RNAlib C-library are required to be linked
against libgsl. If this introduces any problems in your own third-party
programs that link against RNAlib, you can turn off a larger protion of available
minimizers in RNApvmin and linking against libgsl alltogether, using the switch
./configure --without-gslSingle precision partition function
Calculation of partition functions (via RNAfold -p) uses double
precision floats by default, to avoid overflow errors on longer sequences.
If your machine has little memory and you dont't plan to fold sequences
over 1000 bases in length you can compile the package to do the computions
in single precision by running
./configure --enable-floatpf
For a complete list of all ./configure options and important
environment variables, type
./configure --helpFor more general information on the buid process see the
INSTALL.configure file.
Programs
Although the man pages are also included in the ViennaRNA Package itself it is sometimes useful to have an HTML translation making them accessible through a web browser.
| Program | Description |
|---|---|
| AnalyseDists | Analyse a distance matrix |
| AnalyseSeqs | Analyse a set of sequences of common length |
| Kinfold | Simulate kinetic folding of RNA secondary structures |
| kinwalker | Predict RNA folding trajectories |
| RNA2Dfold | Compute MFE structure, partition function and representative sample structures of k,l neighborhoods |
| RNAaliduplex | Predict conserved RNA-RNA interactions between two alignments |
| RNAalifold | Calculate secondary structures for a set of aligned RNA sequences |
| RNAcofold | Calculate secondary structures of two RNAs with dimerization |
| RNAdistance | Calculate distances between RNA secondary structures |
| RNAdos | Calculate the density of states for each energy band of an RNA |
| RNAduplex | Compute the structure upon hybridization of two RNA strands |
| RNAeval | Evaluate free energy of RNA sequences with given secondary structure |
| RNAfold | Calculate minimum free energy secondary structures and partition function of RNAs |
| RNAforester | Compare RNA secondary structures via forest alignment |
| RNAheat | Calculate the specific heat (melting curve) of an RNA sequence |
| RNAinverse | Find RNA sequences with given secondary structure (sequence design) |
| RNALalifold | Calculate locally stable secondary structures for a set of aligned RNAs |
| RNALfold | Calculate locally stable secondary structures of long RNAs |
| RNAlocmin | Calculate local minima from structures via gradient walks |
| RNAmultifold | Compute thermodynamic properties for interaction complexes of multiple RNAs |
| RNApaln | RNA alignment based on sequence base pairing propensities |
| RNApdist | Calculate distances between thermodynamic RNA secondary structures ensembles |
| RNAparconv | Convert energy parameter files from ViennaRNA 1.8 to 2.0 format |
| RNAPKplex | Predict RNA secondary structures including pseudoknots |
| RNAplex | Find targets of a query RNA |
| RNAplfold | Calculate average pair probabilities for locally stable secondary structures |
| RNAplot | Draw RNA Secondary Structures in PostScript, SVG, or GML |
| RNApvmin | Calculate a perturbation vector that minimizes discrepancies between predicted and observed pairing probabilities |
| RNAsnoop | Find targets of a query H/ACA snoRNA |
| RNAsubopt | Calculate suboptimal secondary structures of RNAs |
| RNAup | Calculate the thermodynamics of RNA-RNA interactions |
| RNAxplorer | Explore the RNA conformation space through sampling and other techniques |
Utilities
The Vienna RNA package comes with a number of small utilities, many of them to manipulate the
PostScript files produced by the structure prediction programs RNAfold and RNAalifold.
Most of the Perl utilities contain embedded pod documentation. Type e.g.
perldoc relplot.plfor detailed instructions.
| Tool | Description |
|---|---|
| ct2db | Produce dot bracket notation of an RNA secondary structure given as mfold .ct file |
| b2mt.pl |
Produce a mountain representation of a secondary structure from it's
dot-bracket notation, as produced by RNAfold.
Output consists of simple x y data suitable as input to a plotting program. The
mountain representation is a xy plot with sequence position on the x-axis and the
number of base pairs enclosing that position on the y-axis.
Example: RNAfold < my.seq | b2mt.pl | xmgrace -pipe 
|
| cmount.pl |
Produce a PostScript mountain plot from a color dot plot as created by RNAalifold -p
or alidot -p. Each base pair is represented by a trapez whose color encodes
the number of consistent and compensatory mutations supporting that pair: Red marks pairs
with no sequence variation; ochre, green, turquoise, blue, and violet mark pairs with
2,3,4,5,6 different types of pairs, respectively.
Example: cmount.pl alidot.ps > cmount.ps 
|
| coloraln.pl |
Reads a sequence alignment in CLUSTAL format and a consensus secondary structure (which
it extracts from a secondary structure plot as produced by RNAalifold), and produces a
postscript figure of the alignment annotated using the consensus structure, coloring
base pair using the same color scheme as cmount.pl, RNAalifold and alidot.
Example: coloraln.pl -s alirna.ps file.aln > coloraln.ps 
|
| colorrna.pl |
Reads a consensus secondary structure plot and a color dot plot as produced by RNAalifold -p,
and writes a new secondary structure plot in which base pairs a colored using the color information
from the dot plot.
Example: colorrna.pl alirna.ps alidot.ps > colorRNA.ps |
| mountain.pl |
Similar to b2mt.pl, but produces a mountain plot from the pair probabilities
contained in a PostScript dot plot. It write 3 sets of x y data, suitable as input for a
plot program. The first two sets containing the mountain representation from pair probabilities
and MFE structure, the third set is the "positional entropy" a measure of structural well-definedness.
Example: mountain.pl dot.ps | xmgrace -pipe 
|
| refold.pl | Refold using consensus structure as constraint |
| relplot.pl |
Reads a postscript secondary structure plot and a dot plot containing pair probabilities
as produced by RNAfold -p, and produces a new structure plot, color annotated
with reliability information in the form of either pair probabilities or positional entropy
(default).
Example: relplot.pl foo_ss.ps foo_dp.ps > foo_rss.ps 
|
| rotate_ss.pl |
Reads a postscript secondary structure plot as produced by RNAfold and
produces a new rotated and/or mirrored structure plot.
Example: rotate_ss.pl -a 30 -m foo_ss.ps > foo_new_ss.ps |
| switch.pl |
Design sequences that can adopt two different structure, i.e. design RNA switches.
The program will sample the set of sequences compatible with two input structures
in order to find sequences with desired thermodynamic and kinetic properties. In
particular it is possible to specify
|
| RNAdesign.pl |
Flexible design of multi-stable RNA molecules. An initially random sequence is
iteratively mutated and evaluated according to an objective function (see Option: --optfun).
Whenever a better scoring sequence has been found, the mutation is accepted, the algorithm terminates
once a local minimum is found. This script makes heavy use of the new RNA::Design
Perl 5 sub-package provided by the latest ViennaRNA Package.
|
RNAlib Reference Manual
The core of the ViennaRNA Package is formed by a collection of routines for the prediction and comparison of RNA secondary structures. These routines can be accessed through the stand-alone programs and utilities described above, which should be sufficient for most users. For those who wish to develop their own programs we provide a library that can either be linked to your own C/C++ code, or accessed through our scripting language interface. Currently, we provide support for Perl and Python.
Follow this link to view the HTML version of the RNAlib API Reference Manual.
 The documentation can also be
downloaded as a PDF document
The documentation can also be
downloaded as a PDF document
The documentation can also be found at Read the Docs
Terms and Definitions
Throughout these webpages, we use many terms related to RNA secondary structure prediction. Below you'll find a list of some often used vocabulary and its corresponding definition.
| Term | Description |
|---|---|
| Dot-Bracket notation |
Pseudo-knot free secondary structures can be represented in the
space-efficient bracket notation, which is used throughout
the ViennaRNA package. A structure on a sequence of length n is
represented by a string of equal length consisting of matching
brackets and dots. A base pair between base i and j is represented by
a ( at position i and a ) at position j, unpaired bases are
represented by dots. Thus the secondary structure
(((..((((...)))).)))is equivalent to: 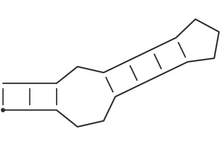
i.e. a stem-loop structure consisting of a an outer helix of 3 base pairs followed by an interior loop of size 3, a second helix of length 4, and a hairpin loop of size 3.
Base pair probabilities are sometimes summarized in pseudo bracket
notation with the additional symbols |
Comments and Bug Reports
If in doubt our program is right, nature is at fault.
Comments and bug reports should be sent to rna@tbi.univie.ac.at