
A short Tutorial on RNA Bioinformatics
The ViennaRNA Package and related Programs
August 2, 2017
RNA Web Services
This tutorial aims to give a basic introduction to using the command line programs in the ViennaRNA Package in a UNIX-like (LINUX) environment. Of course, some of you may ask “Why are there no friendly graphical user interfaces?”. Well, there are some, especially in the form of web services.If a few simple structure predictions is all you want to do, there are several useful sites for doing RNA structure analysis available on the web. Indeed many of the tasks described below can be performed using various web servers.
Useful Web Services
-
Michael Zuker’s
mfoldserver computes (sub)optimal structures and hybridization for DNA and RNA sequences with many options.
Mfold Website -
BiBiServ, several small services e.g. pseudo-knot prediction
pknotsRG, bi-stable structurespaRNAss, alignmentRNAforester, visualizationRNAmovies, suboptimal structuresRNAshapes
Bielefeld Bioinformatics Service -
The ViennaRNA Server offers web access to many tools of the ViennaRNA
Package, e.g.
RNAfold,RNAalifold,RNAinverseand soonRNAz
ViennaRNA Webservices -
several specialized servers such as
-
pfoldconsensus structure prediction
pfold RNA fold server
-
s-foldstochastic suboptimals and siRNA design
Sfold Webservices
-
StrAlprogressiv ncRNA alignment tool
StrAl Webservice
-
Web servers are also a good starting point for novice users since they provide a more intuitive interface. Moreover, the ViennaRNA Server will return the equivalent command line invocation for each request, making the transition from web services to locally installed software easier.
On the other hand, web servers are not ideal for analyzing many or very long sequences and usually they offer only few often-used tasks. Much the same is true for point-and-click graphical interfaces. Command line tools, on the other hand, are ideally suited for automating repetitive tasks. They can even be combined in pipes to process the results of one program with another or they can be used in parallel, running tens or hundreds of tasks simultaneously on a cluster of PCs.
You can try some of these web services in parallel to the exercises below.
Get started
Typographical Conventions
-
Constant width fontis used for program names, variable names and other literal text like input and output in the terminal window. -
Lines starting with a
$within a literal text block are commands. You should type the text following the$into your terminal window finishing by hitting the Enter-key. (The$signifies the command line prompt, which may look different on your system). - All other lines within a literal text block are the output from the command you just typed.
Data Files
Data files containing the sequences used in the examples below are shipped with this tutorial.Terminal, Command line and Editor
- You can get a terminal by moving your mouse-pointer to an empty spot of your desktop, clicking the right mouse-button and choose “Open Terminal” from the pull-down menu.
- You can run commands in the terminal by typing them next to the command line prompt (usually something like $) followed by hitting the Enter-key.
-
To get more information about a command type
manfollowed by the command-name and hitting the Enter-key. Leave the man pages by pressing the q-key. -
Redirect a command’s input and output using the following special
characters:
‘’ ties stdout to stdin
‘’ redirects stdout to stdin
‘’ redirects stdout to a file
Here, stdout stands for standard output, which you can normaly see in the terminal. stdin is it’s counterpart, the standard input. The character ‘’ allows you to pipe the standard output of one program directly as standard input into another program, hence, the programs are chained together.
Below you’ll find a list of some useful core commands available in all Linux terminal.
| Command | Description |
pwd | displays the path to the current working directory |
cd | changes the working directory (initially your “HOME”) |
ls | lists files and directorys in the current (or a specified) directory |
mkdir | creates a directory |
rm | removes a file (add option -r for deleting a folder) |
less | shows file(s) one page at a time |
echo | prints string(s) to standard output |
wc | command prints the number of newlines, words and bytes in a specified file |
For more information regarding these commands prepend --help to the program
call, like this:
Try a few commands on your own, e.g.
Here the stdout from the ls command was written to a file called file_list. The next
command shows the content of file_list. We quit less by pressing the q-key and removing
the file. ls
less pipes the output in the less program without writing it to a file.
Now we create our working directory including subfolders and our first sequence file using the commands we just learned. Have in mind that you create a good structure so you can find your data easily.
First find out in which directory your are in by typing
It should look similar to
To insure yourself, that you are in the correct directory type (˜is the shortcut for the
home-directory)
Now create a new folder in your home directory
Here we created two new folders in our HOME, Tutorial and a subfolder called Data,
then we jumped to the Data-folder and wrote a short DNA sequence to the BAZ.seq
file.
For further processing we need a RNA sequence instead of an DNA sequence, so we
need to replace the T by an U by executing following command using sed (the
stream editor).
$ sed -i 's/T/U/g' BAZ.seq
The program is called via sed, -i tells sed to replace the existing file (in this case
BAZ.seq). s stands for substitute T by U and g tells sed to replace all occuring T’s
in the file globaly).
When we look at our file using less we should see our new sequence
“AUGAAGAUGA”
$ less BAZ.seq
Installing Software from Source
Many bioinformatics programs are available only as source code that has to be
compiled and installed. We’ll demonstrate the standard way to install programs from
source using the ViennaRNA Package.
ViennaRNA PackageYou can either get the required package, depending on which operating system you run (precompiled package is availaible for distinct distributions like Fedora, Arch Linux, Debian, Ubuntu, Windows) or you compile the source code yourself. Here we are compiling the programs ourself. Have a look at the file
INSTALL distributed with the ViennaRNA Package for more detail or read the
documentation on the url. Subsequently the instructions for building the source code are:
-
Go to your
Tutorialsfolder and create a directory$ cd .. $ mkdir downloads $ cd downloads
-
Download the
ViennaRNA Packagefrom http://www.tbi.univie.ac.at/RNA/index.html and save it in to the newly created directory. -
Unpack the gzipped tar archive by running: (Replace [2.1.9] with the actual
version number)
$ tar -zxf ViennaRNA-[2.1.9].tar.gz
-
list the content of the directory
$ ls -F ViennaRNA-[2.1.9]/ ViennaRNA-[2.1.9].tar.gz
Build the ViennaRNA Package
ViennaRNA PackageThe installation location can be controlled through options to the
configure script. E.g. to change the default installation location to
the directory VRP in your $HOME/Tutorial directory use the --prefix tag so the
compiler knows that the target directory is changed.
-
To configure and build the package just run the following commands.
$ cd ViennaRNA-[2.1.9] $ mkdir -p ~/Tutorial/Progs/VRP $ ./configure --prefix=$HOME/Tutorial/Progs/VRP $ make $ make install
You already know the
cdand themkdircommand,./configurechecks whether all dependencies are fulfilled and exits the script if some major requirements are missing. If all is ok it creates theMakefilewhich then is used to start the buildingprocess viamake install. -
To install the
ViennaRNA packagesystem wide (only for people with superuser privileges, which we are NOT!) run$ ./configure $ make $ make install
You find the installed files in
-
$HOME/Tutorial/Progs/VRP/bin(programs) -
$HOME/Tutorial/Progs/VRP/share/ViennaRNA/bin(perl scripts)
Wherever you installed the main programs of the ViennaRNA Package, make sure the
path to the executables shows up in your PATH environment variable. To check the
contents of the PATH environment variable simply run
$ echo $PATH
For easier handling we now create a folder containing all our binaries as well as perl scripts and copy them into a common folder.
$ cd ~/Tutorial/Progs/ $ cp VRP/share/ViennaRNA/bin/* .
Now you can show the contents of the folder using the command ls.
Also copy the binaries from the VRP/bin folder. In the next step we add the path of
the directory to the PATH environment variable (e.g. use pwd) so we don’t need to
write the hole path every time we call it.
$ export PATH=${HOME}/Tutorial/Progs:${PATH} Note that this is only a temporary solution. If you want the path to be
permanently added you need to add the line above to the config file of your shell
environment. Typically bash is the standard. You need to add the export line above
to the .bashrc in your homedirectory. To reload the contents of .bashrc
type
$ source ~/.bashrc
or close the current terminal and open it again. (Remember, this works only for
the bash shell.) To check if everything worked out find which source you
use.
$ which RNAfold
The shown path should point to $HOME/Tutorial/Progs/. Finally try to get a brief
description of a program e.g.
$ RNAfold --help
If this doesn’t work reread the steps described above more carefully.
What’s in the ViennaRNA Package
The core of the ViennaRNA Package is formed by
a collection of routines for the prediction and comparison of RNA secondary
structures. These routines can be accessed through stand-alone programs, such as
RNAfold, RNAdistance etc., which should be sufficient for most users. For those who
wish to develop their own programs a library which can be linked to your own code is
provided.
-
make a directory listing of
downloads/ViennaRNA-2.1.9/$ ls -F ~/Tutorial/downloads/ViennaRNA-2.1.9/
acinclude.m4 config.h.in H/ Makefile Readseq/
aclocal.m4 config.log INSTALL Makefile.am RNAforester/
AUTHORS config.status* INSTALL.configure Makefile.in RNAlib2.pc
ChangeLog config.sub* install-sh* man/ RNAlib2.pc.in
Cluster/ configure* interfaces/ misc/ stamp-h1
compile* configure.ac Kinfold/ missing* THANKS
config/ COPYING lib/ NEWS Utils/
config.guess* depcomp* libsvm-2.91/ Progs/
config.h doc/ m4/ READMEYou now see the contents of the ViennaRNA-2.1.9 folder. Directorys are marked by a ”/” and the
”*” indicates executable files. The Makefile contains the rules to compile the code, the Perl folder
and the Progs folder hold all the binaries but Kinfold and RNAforester and Utils contains the perl
scripts. configure handels distinct options for installation and creation of the Makefile. INSTALL
covers installation instructions and the README file contains information about the ViennaRNA
Package.
| RNA2Dfold | Compute coarse grained energy landscape of representative sample structures |
| RNAaliduplex | Predict conserved RNA-RNA interactions between two alignments |
| RNAalifold | Calculate secondary structures for a set of aligned RNA sequences |
| RNAcofold | Calculate secondary structures of two RNAs with dimerization |
| RNAdistance | Calculate distances between RNA secondary structures |
| RNAduplex | Compute the structure upon hybridization of two RNA strands |
| RNAeval | Evaluate free energy of RNA sequences with given secondary structure |
| RNAfold | Calculate minimum free energy secondary structures and partition function of RNAs |
| RNAheat | Calculate the specific heat (melting curve) of an RNA sequence |
| RNAinverse | Find RNA sequences with given secondary structure (sequence design) |
| RNALalifold | Calculate locally stable secondary structures for a set of aligned RNAs |
| RNALfold | Calculate locally stable secondary structures of long RNAs |
| RNApaln | RNA alignment based on sequence base pairing propensities |
| RNApdist | Calculate distances between thermodynamic RNA secondary structures ensembles |
| RNAparconv | Convert energy parameter files from ViennaRNA 1.8 to 2 format |
| RNAPKplex | Predict RNA secondary structures including pseudoknots |
| RNAplex | Find targets of a query RNA |
| RNAplfold | Calculate average pair probabilities for locally stable secondary structures |
| RNAplot | Draw and markup RNA secondary structures in PostScript, SVG, or GML |
| RNApvmin | Find a vector of perturbation energies which may further be used to constrain folding |
| RNAsnoop | Find targets of a query H/ACA snoRNA |
| RNAsubopt | Calculate suboptimal secondary structures of RNAs |
| RNAup | Calculate the thermodynamics of RNA-RNA interactions |
| Kinfold | simulates the stochastic folding kinetics of RNA sequences into secondary structures |
| RNAforester 1 | compare RNA secondary structures via forest alignment |
| b2ct | converts dot-bracket notation to Zukers mfold ’.ct’ file format |
| b2mt.pl | converts dot-bracket notation to x y values |
| cmount.pl | generates colored mountain plot |
| coloraln.pl | colorize an alirna.ps file |
| colorrna.pl | colorize a secondary structure with reliability annotation |
| ct2b.pl | converts Zukers mfold ’.ct’ file format to dot-bracket notation |
| dpzoom.pl | extract a portion of a dot plot |
| mountain.pl | generates mountain plot |
| popt | extract Zuker’s p-optimal folds from subopt output |
| refold.pl | refold using consensus structure as constraint |
| relplot.pl | add reliability information to a RNA secondary structure plot |
| rotate_ss.pl | rotate the coordinates of an RNA secondary structure plot |
| switch.pl | describes RNA sequences that exhibit two almost equally stable structures |
All programs that are shipped with the ViennaRNA Package provide some documentation in the form
of “man pages”. In UNIX like environments, these manual pages can be viewed using the man
command after successfully installing the ViennaRNA Package:
$ man RNAalifold
Alternatively, an online version of the manual pages is available at
https://www.tbi.univie.ac.at/RNA/documentation.html#programs. Note, that the MANPATH
environment variable requires to be updated if the ViennaRNA Package has been installed in a
non-standard path.
There also is a helpful documentation in the folder of the ViennaRNA Package: /Tutorial/downloads/ViennaRNA-2.1.9/doc/RNAlib-2.1.9.pdf
Most Perl scripts carry embedded documentation that is displayed by typing
$ perldoc coloraln.pl
in the folder where the script is located. All scripts and programs give short usage instructions when
called with the -h command line option (e.g. RNAalifold -h).
The Input File Format
RNA sequences come in a variety of formats. The sequence format used throughout the ViennaRNA Package is very simple. An sequence file contains one or more sequences. Each sequence must appear as a single line in the file without embedded white spaces. A sequence may be preceded by a special line starting with the ‘>’ character
followed by a sequence name. This name will be used by the programs in the ViennaRNA
Package as basename for the PostScript output files for this sequence. Note that this
is almost the fasta sequence format, except that no line-breaks are allowed within a
sequence while the header line is optional. Following programs provide full fasta support:
RNAfold, RNAsubopt, RNAcofold, RNAKplex, RNALfold, RNAplfold, RNAeval, RNAplot,
RNAheat
Structure Prediction on single Sequences
The Program RNAfold
Our first task will be to do a structure prediction using RNAfold. This
should get you familiar with the input and output format as well as the graphical output
produced.
RNAfold reads RNA sequences from stdin, calculates their minimum free energy (MFE) structure,
prints the MFE structure in dot-bracket notation and its free energy to stdout. If the -p option is set
it also computes the partition function, the base pairing probability matrix and additionally prints
the free energy of the thermodynamic ensemble, the frequency of the MFE structure in the ensemble
and the ensemble diversity to stdout. Another useful option is the --MEA option, which also shows
the maximum expected accuracy, but remember that this also needs more CPU time than without
--MEA.
-
Use a text editor (emacs, vi, nano, gedit) to prepare an input file by pasting the text below
and save it under the name
test.seqin yourDatafolder.> test CUACGGCGCGGCGCCCUUGGCGA
-
Compute the best (MFE) structure for this sequence
$ RNAfold < test.seq CUACGGCGCGGCGCCCUUGGCGA ...........((((...)))). ( -5.00)
The last line of the text output contains the predicted MFE structure as dot-bracket notation and
its free energy in kcal/mol. A dot in the dot-bracket notation represents an unpaired position,
while a base pair (i, j) is represented by a pair of matching parentheses at position i and
j.
RNAfold created a file named test_ss.eps. The filename is taken from the fasta header; if there’s no
header the output is simply called rna.eps.
Let’s take a look at the output file with gv, a
PostScript2
viewer. The & at the end starts the program in the background.
$ gv test_ss.ps &
Compare the dot-bracket notation to the PostScript drawing shown in the file test_ss.eps.
The calculation above does not tell us whether the predicted structure is the only possibility or not, so let’s look at the equilibrium ensemble instead.
-
Run
RNAfold -p --MEAto compute the partition function and pair probabilities as well as the maximum expected accurarcy. -
Look at the generated PostScript files
test_ss.epsandtest_dp.eps$ RNAfold -p --MEA < test.seq CUACGGCGCGGCGCCCUUGGCGA ...........((((...)))). ( -5.00) ....{,{{...||||...)}}}. [ -5.72] ....................... { 0.00 d=4.66} ......((...))((...))... { 2.90 MEA=14.79} frequency of mfe structure in ensemble 0.311796; ensemble diversity 6.36
Here the last four lines are new compared to the text output without the -p --MEA options. The
partition function is a rough measure for the well-definedness of the MFE structure. The third line
shows a condensed representation of the pair probabilities of each nucleotide, similar to the
dot-bracket notation, followed by the ensemble free energy (-kT ln(Z)) in kcal/mol. The next two
lines represent the centroid structure with its free energy, its distance to the ensemble and the MEA.
The last line shows the frequency of the MFE structure in the ensemble of secondary structures and
the diversity of the ensemble.
”.” denotes bases that are essentially unpaired, ”,” weakly paired,
””strongly paired without preference,
”{},()” weakly (33%) upstream
(downstream) paired or strongly (66%)
up-/downstream paired bases, respectively.
Note that the MFE structure is adopted only with 31% probability, also the diversity is very high for
such a short sequence.
For rotating the secondary structure plot there is a usefull tool called rotate_ss.pl included in the
ViennaRNA Package. Just read the perldoc for this tool to know how to handle the rotation and
use the information to get your secondary structure in a vertical position.
$ perldoc rotate_ss.pl
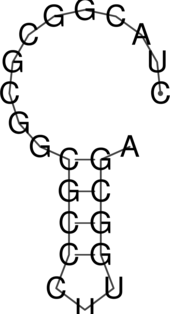
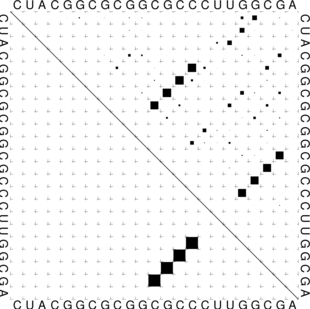
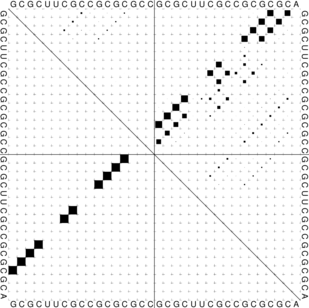
The “dot plot” in test_dp.eps shows the pair probabilities within the equilibrium ensemble as
matrix, and is an excellent way to visualize structural alternatives. A square at row
and column
indicates a
base pair. The area of a square in the upper right half of the matrix is proportional to the probability of
the base pair
within the equilibrium ensemble. The lower left half shows all pairs belonging to the MFE structure.
While the MFE consists of a single helix, several different helices are visualized in the pair
probabilities.
Next, let’s use the relplot utility to visualize which parts of a predicted MFE are well-defined and
thus more reliable. Also let’s use a real example for a change and produce yet another representation
of the predicted structure, the mountain plot.
Fold the 5S rRNA sequence and visualize the structure. (The
5S.seq
is shipped with the tutorial)
$ RNAfold -p < 5S.seq $ mountain.pl 5S_dp.ps | xmgrace -pipe $ relplot.pl 5S_ss.ps 5S_dp.ps > 5S_rss.ps
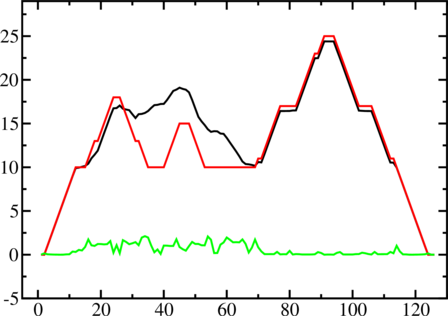
A mountain plot is especially useful for long sequences where conventional structure drawings
become terribly cluttered. It is a xy-diagram plotting the number of base pairs enclosing a sequence
position versus the position. The Perl script mountain.pl transforms a dot plot into the
mountain plot coordinates which can be visualized with any xy-plotting program, e.g.
xmgrace.
The resulting plot shows three curves, two mountain plots derived from the MFE structure (red) and
the pairing probabilities (black) and a positional entropy curve (green). Well-defined regions are
identified by low entropy. By superimposing several mountain plots structures can easily be
compared.
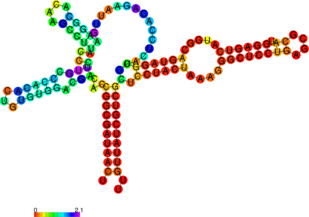
The perl script relplot.pl adds reliability information to a RNA secondary structure plot in the form
of color annotation. The script computes a well-definedness measure we call “positional entropy”
( for
those who want to know the details) and encodes it as color hue, ranging from red (low entropy,
well-defined) via green to blue and violet (high entropy, ill-defined). In the example above two
helices of the 5S RNA are well-defined (red) and indeed predicted correctly, the left arm is not quite
correct and disordered.
For the figure above we had to rotate and mirror the structure plot, e.g.
$ rotate_ss.pl -a 180 -m 5S_rss.ps > 5S_rot.ps
You can manually add annotation to structure drawings using the RNAplot program (for
information see the man page). Here’s a somewhat complicated example:
$ RNAfold < 5S.seq > 5S.fold $ RNAplot --pre "76 107 82 102 GREEN BFmark 44 49 0.8 0.8 0.8 Fomark \ 1 15 8 RED omark 80 cmark 80 -0.23 -1.2 (pos80) Label 90 95 BLUE Fomark" < 5S.fold $ gv 5S_ss.ps
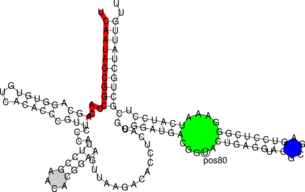
RNAplot is a very useful tool to color plots. The --pre tag adds PostScript code to color distinct
regions of your molecule. There are some predefined statements with different options for
annotations listed below:
i cmark | draws circle around base i |
i j c gmark | draw basepair i,j with c counter examples in grey |
i j lw rgb omark | stroke segment i...j with linewidth lw and color (rgb) |
i j rgb Fomark | fill segment i...j with color (rgb) |
i j k l rgb BFmark | fill block between pairs i,j and k,l with color (rgb) |
i dx dy (text) Label | adds a textlabel with an offset dx and dy relative to base i |
Predefined color options are BLACK, RED, GREEN, BLUE, WHITE but you can also replace the value to
some standard RGB code (e.g. 0 5 8 for lightblue).
To see what exactly the alternative structures of our sequence are, we need to predict suboptimal structures.
In order to further improve the quality of secondary structure predictions, mapping experiments like SHAPE (selective 2’-hydroxyl acylation analyzed by primer extension) can be used to exerimentally determine the pairing status for each nucleotide. In addition to thermodynamic based secondary structure predictions, RNAfold supports the incorporation of this additional experimental data as soft constraints.
If you want to use SHAPE data to guide the folding process, please make sure that your experimental data is present in a text file, where each line stores three white space separated columns containing the position, the abbreviation and the normalized SHAPE reactivity for a certain nucleotide.
1 G 0.134
2 C 0.044
3 C 0.057
4 G 0.114
5 U 0.094
...
...
...
71 C 0.035
72 G 0.909
73 C 0.224
74 C 0.529
75 A 1.475The second column, which holds the nucleotide abbreviation, is optional. If it is present, the data will be used to perform a cross check against the provided input sequence. Missing SHAPE reactivities for certain positions can be indicated by omitting the reactivity column or the whole line. Negative reactivities will be treated as missing. Once the SHAPE file is ready, it can be used to constrain folding:
$ RNAfold --shape=rna.shape --shapeMethod=D < rna.seq
The Program RNApvmin
The program RNApvmin reads a RNA sequence from stdin and uses an iterative minimization process
to calculate a perturbation vector that minimizes the discripancies between predicted pairing
probabilites and observed pairing probabilities (deduced from given shape reactivities). The
experimental SHAPE data has to be present in the file format described above. The application will
write the calculated vector of perturbation energies to stdout, while the progress of the minimization
process is written to stderr. The resulting perturbation vector can be interpreted directly and gives
usefull insights into the discrepancies between thermodynamic prediction and experimentally
determined pairing status. In addition the perturbation energies can be used to constrain folding
with RNAfold:
$ RNApvmin rna.shape < rna.seq >vector.csv $ RNAfold --shape=vector.csv --shapeMethod=W < rna.seq
The perturbation vector file uses the same file format as the SHAPE data file. Instead of SHAPE reactivities the raw perturbation energies will be storred in the last column. Since the energy model is only adjusted when necessary, the calculated perturbation energies may be used for the interpretation of the secondary structure prediction, since they indicate which positions require major energy model adjustments in order to yield a prediction result close to the experimental data. High perturbation energies for just a few nucleotides may indicate the occurrence of features, which are not explicitly handled by the energy model, such as posttranscriptional modifications and intermolecular interactions.
The Program RNAsubopt
RNAsubopt calculates all suboptimal secondary structures within a given
energy range above the MFE structure. Be careful, the number of structures returned grows
exponentially with both sequence length and energy range.
-
Generate all suboptimal structures within a certain energy range from the
MFEspecified by the-eoption.$ RNAsubopt -e 1 -s < test.seq CUACGGCGCGGCGCCCUUGGCGA -500 100 ...........((((...)))). -5.00 ....((((...))))........ -4.80 (((.((((...))))..)))... -4.20 ...((.((.((...)).)).)). -4.10
The text output shows an energy sorted list (option -s) of all secondary structures within
1 kcal/mol of the MFE structure. Our sequence actually has a ground state structure (-5.70) and
three structures within 1 kcal/mol range.
MFE folding alone gives no indication that there are actually a number of plausible structures.
Remember that RNAsubopt cannot automatically plot structures, therefore you can use the
tool RNAplot. Note that you CANNOT simply pipe the output of RNAsubopt to RNAplot
using
$ RNAsubopt < test.seq | RNAplot
You need to manually create a file for each structure you want to plot. Here, for example we created a new file named suboptstructure.txt:
> suboptstructure-4.20 CUACGGCGCGGCGCCCUUGGCGA (((.((((...))))..)))...
The fasta header is optional, but useful (without it the outputfile will be named rna.ps). The next two lines contain the sequence and the suboptimal structure you want to plot; in this case we plotted the structure with the folding energy of -4.20. Then plot it with
$ RNAplot < suboptstructure.txt
Note that the number of suboptimal structures grows exponentially with sequence length and
therefore this approach is only tractable for sequences with less than 100 nt. To keep the number of
suboptimal structures manageable the option --noLP can be used, forcing RNAsubopt to produce only
structures without isolated base pairs. While RNAsubopt produces all structures within an energy
range, mfold produces only a few, hopefully representative, structures. Try folding the sequence on
the mfold server at
http://mfold.rna.albany.edu/?q=mfold.
Sometimes you want to get information about unusual properties of the Boltzmann ensemble (the sum of all RNA structures possible) for which no specialized program exists. For example you want to know all fractions of a bacterial mRNA in the Boltzmann ensemble where the Shine-Dalgarno (SD) sequence is unpaired. If the SD sequence is concealed by secondary structure the translation efficiency is reduced.
In such cases you can resort to drawing a representative sample of structures from the Boltzmann
ensemble by using the option -p. Now you can simply count how many structures in the sample
possess the feature you are looking for. This number divided by the size of your sample gives you the
desired fraction.
The following example calculates the fraction of structures in the ensemble that have bases 6 to 8 unpaired.
- Draw a sample of size 10,000 from the Boltzmann ensemble
- Calculate the desired property by using a perl script
$ RNAsubopt -p 10000 < test.seq > tt
$ perl -nle '$h++ if substr($_,5,3) eq "...";
END {print $h/$.}' tt
0.391960803919608A far better way to calculate this property is to use RNAfold -p to get
the ensemble free energy, which is related to the partition function via
, for the unconstrained
() and the
constrained case (),
where the three bases are not allowed to form base pairs (use option -C), and evaluate
to get
the desired probability.
So let’s do the calculation using RNAfold.
$RNAfold -p
Input string (upper or lower case); @ to quit
....,....1....,....2....,....3....,....4....,....5....,....6....,....7....,....8
CUACGGCGCGGCGCCCUUGGCGA
length = 23
CUACGGCGCGGCGCCCUUGGCGA
...........((((...)))).
minimum free energy = -5.00 kcal/mol
....{,{{...||||...)}}}.
free energy of ensemble = -5.72 kcal/mol
....................... { 0.00 d=4.66}
frequency of mfe structure in ensemble 0.311796; ensemble diversity 6.36 Now we have calculated the free ensemble energy of the ensemble over all structures (F_u), in the
next step we have to calculate it for the structures using a constraint(F_c).
Following notation has to be used for defining the constraint:
- : paired with another base
- . : no constraint at all
- x : base must not pair
- : base i is paired with a base j¡i
- : base i is paired with a base j¿i
-
matching brackets ( ): base i pairs base j
So our constraint should look like this:
.....xxx...............
Next call the application with following command and provide the sequence and constraint we just created.
$ RNAfold -p -C
The output should look like this
length = 23
CUACGGCGCGGCGCCCUUGGCGA
...........((((...)))).
minimum free energy = -5.00 kcal/mol
...........((((...)))).
free energy of ensemble = -5.14 kcal/mol
...........((((...)))). { -5.00 d=0.42}
frequency of mfe structure in ensemble 0.792925; ensemble diversity 0.79Afterwards evaluate the desired probability according to the formula given before e.g. with a simple perl script.
$ perl -e 'print exp(-(5.72-5.14)/(0.00198*310.15))."\n"'
You can see that there is a slight difference between the RNAsubopt run with 10,000 samples and the
RNAfold run including all structures.
RNA folding kinetics
RNA folding kinetics describes the dynamical process of how a RNA molecule approaches to its unique folded biological active conformation (often referred to as the native state) starting from an initial ensemble of disordered conformations e.g. the unfolded open chain. The key for resolving the dynamical behavior of a folding RNA chain lies in the understanding of the ways in which the molecule explores its astronomically large free energy landscape, a rugged and complex hyper-surface established by all the feasible base pairing patterns a RNA sequence can form. The challenge is to understand how the interplay of formation and break up of base pairing interactions along the RNA chain can lead to an efficient search in the energy landscape which reaches the native state of the molecule on a biologically meaningful time scale.RNA2Dfold
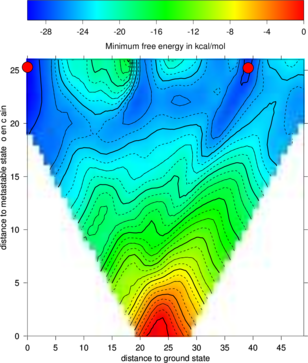
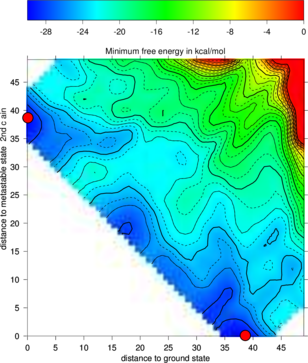
RNA2Dfold is a tool for computing the MFE structure, partition function and representative sample structures of , neighborhoods and projects an high dimensional energy landscape of RNA into two dimensions. Therefore a sequence and two user-defined reference structures are expected by the program. For each of the resulting distance class, the MFE representative, the Boltzmann probabilities and the Gibbs free energy is computed. Additionally, representative suboptimal secondary structures from each partition can be calculated.$ RNA2Dfold -p < 2dfold.inp > 2dfold.out
The outputfile 2dfold.out should look like below, check it out using less.
CGUCAGCUGGGAUGCCAGCCUGCCCCGAAAGGGGCUUGGCGUUUUGGUUGUUGAUUCAACGAUCAC ((((((((((....)))))..(((((....))))).)))))...(((((((((...))))))))). (-30.40) ((((((((((....)))))..(((((....))))).)))))...(((((((((...))))))))). (-30.40) .................................................................. ( 0.00) free energy of ensemble = -31.15 kcal/mol k l P(neighborhood) P(MFE in neighborhood) P(MFE in ensemble) MFE E_gibbs MFE-structure 0 24 0.29435909 1.00000000 0.29435892 -30.40 -30.40 ((((((((((....)))))..(((((....))))).)))))...(((((((((...))))))))). 1 23 0.17076902 0.47069889 0.08038083 -29.60 -30.06 ((((((((((....)))))..(((((....))))).)))))....((((((((...)))))))).. 2 22 0.03575448 0.37731068 0.01349056 -28.50 -29.10 ((((.(((((....)))))..(((((....)))))..))))....((((((((...)))))))).. 2 24 0.00531223 0.42621709 0.00226416 -27.40 -27.93 ((((((((((....))))...(((((....)))))))))))...(((((((((...))))))))). 3 21 0.00398349 0.29701636 0.00118316 -27.00 -27.75 .(((.(((((....)))))..(((((....)))))..))).....((((((((...)))))))).. 3 23 0.00233909 0.26432372 0.00061828 -26.60 -27.42 ((((((((((....))))...(((((....)))))))))))....((((((((...)))))))).. [...]
For visualizing the output the ViennaRNA Package includes two scripts 2Dlandscape_pf.gri,
2Dlandscape_mfe.gri located in VRP/share/ViennaRNA/. gri (a language for scientific graphics
programing) is needed to create a colored postscript plot. We use the partition function script to
show the free energies of the distance classes (graph below, left):
$ gri ../Progs/VRP/share/ViennaRNA/2Dlandscape_pf.gri 2dfold.out
Compare the output file with the colored plot and determine the MFE minima with
corresponding distance classes. For easier comparision the outputfile of RNA2Dfold can be
sorted by a simple sort command. For further information regarding sort use the --help
option.
$ sort -k6 -n 2dfold.out > sort.out
Now we choose the structure with the lowest energy besides our startstructure, replace the open chain structure from our old input with that structure and repeat the steps above with our new values
-
run
RNA2Dfold -
plot it using
2Dlandscape_pf.gri
The new projection (right graph) shows the two major local minima which are separated by 39 bp
(red dots in figure below) and both are likely to be populated with high probability. The
landscape gives an estimate of the energy barrier separating the two minima (about -20
kcal/mol).
The red dots mark the distance from open chain to the MFE structure respectively the
distance from the 2nd best structure to the MFE. Note that the red dots were manually
added to the image afterwards so don’t panic if you don’t see them in your gri output.
barriers & treekin
The following assumes you have the barriers and treekin programs installed. If not, the current release can be found at http://www.tbi.univie.ac.at/RNA/Barriers/. Installation proceeds as shown for the ViennaRNA Package in section 2.4. One problem that often occurs during treekin installation is the dependency onblas and lapack packages which is not carefully
checked. For further information according to the barriers and treekin program also see the
website.
- Get the barriers source from http://www.tbi.univie.ac.at/RNA/Barriers/
-
extract the archive and go to the directory
$ tar -xzf Barriers-1.5.2.tar.gz $ cd Barriers-1.5.2
-
use the
--prefixoption to install in yourProgsdirectory$ ./configure --prefix=$HOME/Tutorial/Progs/barriers-1.5.2
-
make install
$ make $ make install
Now barriers is ready to use. Apply the same steps to install treekin. Note: Copy the barriers and
treekin binaries to your bin folder or add the path to your PATH variable.
$ echo UCCACGGCUGUUAGUGGAUAACGGC | RNAsubopt --noLP -s -e 10 > barseq.sub $ barriers -G RNA-noLP --bsize --rates < barseq.sub > barseq.bar
You can restrict the number of local minima using the barriers command-line option
--max followed by a number. The option -G RNA-noLP instructs barriers that the input
consists of RNA secondary structures without isolated basepairs. --bsize adds size of the
gradient basins and --rates tells barriers to compute rates between macro states/basins for
use with treekin. Another useful options is --minh to print only minima with a barrier
. Look
at the output file less -S barseq.bar. Use the arrow keys to navigate.
UCCACGGCUGUUAGUGGAUAACGGC 1 (((((........)))))....... -6.90 0 10.00 115 0 -7.354207 23 -7.012023 2 ......(((((((.....))))))) -6.80 1 9.30 32 58 -6.828221 38 -6.828218 3 (((...(((...))))))....... -0.80 1 0.90 1 10 -0.800000 9 -1.075516 4 ....((..((((....)))).)).. -0.80 1 2.70 5 37 -0.973593 11 -0.996226 5 ......................... 0.00 1 0.40 1 14 -0.000000 26 -0.612908 6 ......(((....((.....))))) 0.60 2 0.40 1 22 0.600000 3 0.573278 7 ......((((((....)))...))) 1.00 1 1.50 1 95 1.000000 2 0.948187 8 .((....((......)).....)). 1.40 1 0.30 1 30 1.400000 2 1.228342
The first row holds the input sequence, the successive list the local minima ascending in energy. The meaning of the first 5 columns is as follows
- label (number) of the local minima (1=MFE)
- structure of the minimum
- free energy of the minimum
-
label of deeper local minimum the current minimum merges with (note that the
MFEhas no deeper local minimum to merge with) - height of the energy barrier to the local minimum to merge with
- numbers of structures in the basin we merge with
- number of basin which we merge to
- free energy of the basin
- number of structures in this basin using gradient walk
- gradient basin (consisting of all structures where gradientwalk ends in the minimum)

barriers produced two additional files, the PostScript file tree.eps which represents the
basic information of the barseq.bar file visually (look at the file e.g. gv tree.eps) and a
text file rates.out which holds the matrix of transition probabilities between the local
minima.
The program
treekin is used to simulate the evolution over time of
the population densities of local minima starting from an initial population density distribution
(given
on the command-line) and the transition rate matrix in the file rates.out.
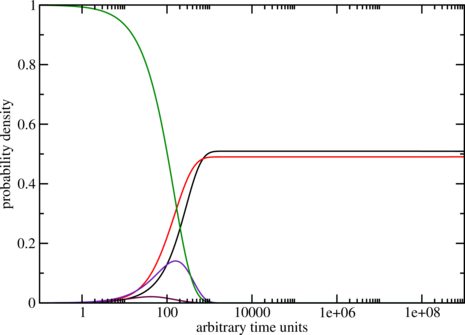
$ treekin -m I --p0 5=1 < barseq.bar | xmgrace -log x -nxy -
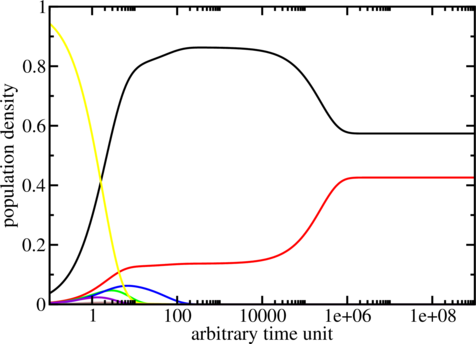
The simulation starts with all the population density in the open chain (local minimum 5, see
barseq.bar). Over time the population density of this state decays (yellow curve) and other local
minima get populated. The simulation ends with the population densities of the thermodynamic
equilibrium in which the MFE (black curve) and local minimum 2 (red curve) are the
only ones populated. (Look at the dot plot of the sequence created with RNAsubopt and
RNAfold!)
Sequence Design
The Program RNAinverse
RNAinverse searches for sequences folding into a predefined structure,
thereby inverting the folding algorithm. Input consists of the target structures (in dot-bracket
notation) and a starting sequence, which is optional.Lower case characters in the start sequence indicate fixed positions, i.e. they can be used to add
sequence constraints. ’N’s in the starting sequence will be replaced by a random nucleotide. For
each search the best sequence found and its Hamming distance to the start sequence are
printed to stdout. If the the search was unsuccessful a structure distance to the target is
appended.
By default the program stops as soon as it finds a sequence that has the target as MFE structure. The
option -Fp switches RNAinverse to the partition function mode where the probability of the target
structure
is maximized. This tends to produce sequences with a more well-defined structure. This probability
is written in dot-brackets after the found sequence and Hamming distance. With the option -R you
can specify how often the search should be repeated.
-
Prepare an input file
inv.incontaining the target structure and sequence constraints(((.(((....))).))) NNNgNNNNNNNNNNaNNN
-
Design sequences using RNAinverse
$ RNAinverse < inv.in GGUgUUGGAUCCGAaACC 5 $ RNAinverse -R5 -Fp < inv.in GGUgUGAACCCUCGaACC 5 GGCgCCCUUUUGGGaGCC 12 (0.967418) CUCgAUCUCACGAUaGGG 6 GGCgCCCGAAAGGGaGCC 13 (0.967548) GUUgAGCCCAUGCUaAGC 6 GGCgCCCUUAUGGGaGCC 10 (0.967418) CGGgUGUUGUGACAaCCG 5 GCGgGUCGAAAGGCaCGC 12 (0.925482) GCCgUAUCCGGGUGaGGC 6 GGCgCCCUUUUGGGaGCC 13 (0.967418)
The output consists of the calculated sequence and the number of mutations needed to get the
MFE-structure from the start sequence (start sequence not shown). Additionaly, with the partition
function folding (-Fp) set, the second output is another refinement so that the ensemble
preferes the MFE and folds into your given structure with a distinct probability, shown in
brackets.
Another useful program for inverse folding is RNA designer, see http://www.rnasoft.ca/. RNA
Designer takes a secondary structure description as input and returns an RNA strand that is likely
to fold in the given secondary structure.
The sequence design application of the ViennaRNA Design Webservices, see
http://nibiru.tbi.univie.ac.at/rnadesign/index.html uses a different approach, allowing for
more than one secondary structure as input. For more detail read the online Documentation and the
next section of this tutorial.
switch.pl
Theswitch.pl script can be used to design bi-stable structures, i.e. structures with two
almost equally good foldings. For two given structures there are always a lot of sequences compatible
with both structures. If both structures are reasonably stable you can find sequences where both
target structures have almost equal energy and all other structures have much higher energies.
Combined with RNAsubopt, barriers and treekin, this is a very useful tool for designing
RNAswitches. The input requires two structures in dot-bracket notation and additionally you can add a sequence.
It is also possible to calculate the switching function at two different temperatures with option -T
and -T2.
Now we try to create an RNA switch using
switch.pl. First we create our
inputfile, then invoke the program using ten optimization runs (-n 10) and do not allow lonely pairs.
Write it out to switch.out
switch.in
((((((((......))))))))....((((((((.......))))))))
((((((((((((((((((........)))))))))))))))))).....
$ switch.pl -n 10 --noLP < switch.in > switch.outswitch.out should look similar like this, the first block represents our bi-stable structures in random
order, the second block shows the resulting sequences ordered by their score.
$ less switch.out GGGUGGACGUUUCGGUCCAUCCUUACGGACUGGGGCGUUUACCUAGUCC 0.9656 CAUUUGGCUUGUGUGUCGAAUGGCCCCGGUACGUAGGCUAAAUGUACCG 1.2319 GGGGGGUGCGUUCACACCCCUCAUUUGGUGUGGAUGUGCUUUCUACACU 1.1554 [...] the resulting sequences are: CAUUUGGCUUGUGUGUCGAAUGGCCCCGGUACGUAGGCUAAAUGUACCG 1.2319 GGGGGGUGCGUUCACACCCCUCAUUUGGUGUGGAUGUGCUUUCUACACU 1.1554 CGGGUUGUAACUGGAUAGCCUGGAAACUGUUUGGUUGUAAUCCGAACAG 1.0956 [...]
Given all 10 suggestions in our switch.out, we select the one with the best score with
some command line tools to use it as an RNAsubopt input file and build up the barriers
tree.
$ tail -10 switch.out | awk '{print($1)}' | head -n 1 > subopt.in
$ RNAsubopt --noLP -s -e 25 < subopt.in > subopt.out
$ barriers -G RNA-noLP --bsize --rates --minh 2 --max 30 < subopt.out > barriers.outtail -10 cuts the last 10 lines from the switch.out file and pipes them into an awk script. The
function print($1) echoes only the first column and this is piped into the head program where the
first line, which equals the best scored sequence, is taken and written into subopt.in. Then
RNAsubopt is called to process our sequence and write the output to another file which is the input
for the barriers calculation.
Below you find an example of the barriertree calculation above done with the right settings
(connected root) on the left side and the wrong RNAsubobt -e value on the right. Keep in mind that
switch.pl performs an stochastic search and the output sequences are different every time because
there are a lot of sequences which fit the structure and switch calculates a new one everytime.
Simply try to make sure.

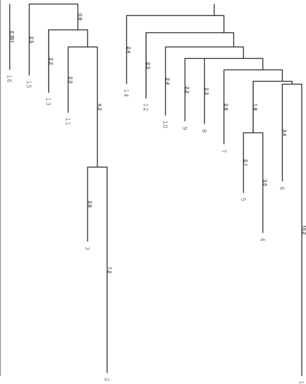 left: Barriers tree as it should look like, all
branches connected to the main root right: disconnected tree due to a too low
left: Barriers tree as it should look like, all
branches connected to the main root right: disconnected tree due to a too low energy range (-e) parameter set in
RNAsubopt.
Be careful to set the range -e high enough, otherwise we get a problem when calculation the kinetics
using treekin. Every branch should be somehow connected to the main root of the tree. Try -e 20
and -e 30 to see the difference in the trees and choose the optimal value. By using --max 30 we
shorten our tree to focus only on the lowest minima. We then select a branch preferably outside of
the two main branches, here branch 30 (may differ from your own calculation). Look at the
barrier tree to find the best branch to start and replace 30 by the branch you would
choose. Now use treekin to plot concentration kinetics and think about the graph you just
created.
$ treekin -m I --p0 30=1 < barriers.out > treekin.out $ xmgrace -log x -nxy treekin.out
The graph could look like the one below, remember everytime you use switch.pl it can
give you different sequences so the output varies too. Here the one from the example.
RNA-RNA Interactions
A common problem is the prediction of binding sites between two RNAs, as in the case of miRNA-mRNA interactions. Following tools of theViennaRNA Package can be used to
calculate base pairing probabilities.
The Program RNAcofold
RNAcofold works much like RNAfold but uses two RNA sequences as input
which are then allowed to form a dimer structure. In the input the two RNA sequences should be
concatenated using the ‘&’ character as separator. As in RNAfold the -p option can be used to
compute partition function and base pairing probabilities.Since dimer formation is concentration dependent, RNAcofold can be used to compute equilibrium
concentrations for all five monomer and (homo/hetero)-dimer species, given input concentrations for
the monomers (see the man page for details).
-
Prepare a sequence file (
t.seq) for input that looks like this>t GCGCUUCGCCGCGCGCC&GCGCUUCGCCGCGCGCA
-
Compute the
MFEand the ensemble properties -
Look at the generated PostScript files
t_ss.psandt_dp.ps
$ RNAcofold -p < t.seq
>t
GCGCUUCGCCGCGCGCC&GCGCUUCGCCGCGCGCA
((((..((..((((...&))))..))..))))... (-17.70)
((((..{(,.((((,,.&))))..}),.)))),,. [-18.26]
frequency of mfe structure in ensemble 0.401754 , delta G binding= -3.95
Concentration Dependency
Cofolding is an intermolecular process, therefore whether duplex formation will actually occur is concentration dependent. Trivially, if one of the molecules is not present, no dimers are going to be formed. The partition functions of the molecules give us the equilibrium constants:with these and mass conservation, the equilibrium concentration of homodimers, heterodimers and
monomers can be computed in dependence of the start concentrations of the two molecules.
This is most easily done by creating a file with the initial concentrations of molecules
and
in two
columns:
-
Prepare a concentration file for input with this little perl script
$ perl -e '$c=1e-07; do {print "$c\t$c\n"; $c*=1.71;} while $c<0.2' > concfileThis script creates a file displaying values from 1e-07 to just below 0.2, with 1.71-fold steps in between. For convenience, concentration of molecule A is the same as concentration of molecule B in each row. This will facilitate visualization of the results.
-
Compute the
MFE, the ensemble properties and the concentration dependency of hybridization.$ RNAcofold -f concfile < t.seq > cofold.out
-
Look at the generated output with
$ less cofold.out
[...] Free Energies: AB AA BB A B -18.261023 -17.562553 -18.274376 -7.017902 -7.290237 Initial concentrations relative Equilibrium concentrations A B AB AA BB A B 1e-07 1e-07 0.00003 0.00002 0.00002 0.49994 0.49993 [...]
The five different free energies were printed out first, followed by a list of all the equilibrium
concentrations, where the first two columns denote the initial (absolute) concentrations of molecules
and
,
respectively. The next five columns denote the equilibrium concentrations of dimers and monomers,
relative to the total particle number. (Hence, the concentrations don’t add up to one, except in the
case where no dimers are built – if you want to know the fraction of particles in a dimer, you have to
take the relative dimer concentrations times 2).
Since relative concentrations of species depend on two independent values - initial concentration of
A as well as initial concentration of B - it is not trivial to visualize the results. For this reason we
used the same concentration for A and for B. Another possibility would be to keep the initial
concentration of one molecule constant. As an example we show the following plot of
. Now
we use some commandline tools to render our plot. We use tail -n +11 to show all lines
starting with line 11 (1-10 are cut) and pipe it into an awk command, which prints every
column but the first from our input. This is then piped to xmgrace. With -log x -nxy
- we tell it to plot the x axis in logarithmic scale and to read data file in X Y1 Y2 ...
format.
$ tail -n +11 cofold.out | awk '{print $2, $3, $4, $5, $6, $7}' | xmgrace -log x -nxy -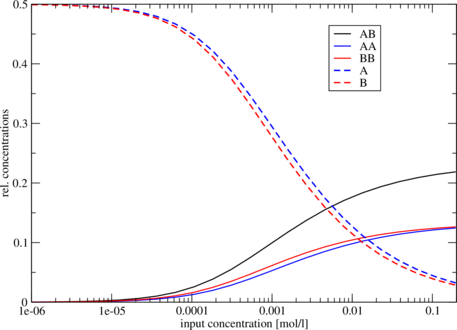
Finding potential binding sites with RNAduplex
If the sequences are very long (many kb)RNAcofold is too slow to be useful. The RNAduplex
program is a fast alternative, that works by predicting only intermolecular base pairs.
It’s almost as fast as simple sequence alignment, but much more accurate than a BLAST
search.
The example below searches the 3’ UTR of an mRNA for a miRNA binding site.
The file
duplex.seq contains the 3’UTR of NM_024615 and
the microRNA mir-145.
$ RNAduplex < duplex.seq >NM_024615 >hsa-miR-145 .(((((.(((...((((((((((.&)))))))))))))))))). 34,57 : 1,19 (-21.90)
Most favorable binding has an interaction energy of -21.90 kcal/mol and pairs up on positions 34-57
of the UTR with positions 1-22 of the miRNA.
RNAduplex can also produce alternative binding sites, e.g. running RNAduplex -e 10 would list all
binding sites within 10 kcal/mol of the best one.
Since RNAduplex forms only intermolecular pairs, it neglects the competition between intramolecular
folding and hybridization. Thus, it is recommended to use RNAduplex as a pre-filter and analyse good
RNAduplex hits additionally with RNAcofold or RNAup. Using the example above, running RNAup will
yield:
$ RNAup -b < duplex.seq >NM_024615 >hsa-miR-145 (((((((&))))))) 50,56 : 1,7 (-8.41 = -9.50 + 0.69 + 0.40) GCUGGAU&GUCCAGU RNAup output in file: hsa-miR-145_NM_024615_w25_u1.out
The free energy of the duplex is -9.50 kcal/mol and shows a discrepancy to the structure and energy
value computed by RNAduplex (differences may arise from the fact that RNAup computes partition
functions rather than optimal structures). However, the total free energy of binding is less favorable
(-8.41 kcal/mol), since it includes the energetic penalty for opening the binding site on the mRNA
(0.69 kcal/mol) and miRNA (0.40 kcal/mol). The -b option includes the probability of unpaired
regions in both RNAs.
You can also run RNAcofold on the example to see the complete structure after hybridization
(neither RNAduplex nor RNAup produce structure drawings). Note however, that the input format for
RNAcofold is different. An input file suitable for RNAcofold has to be created from the duplex.seq
file first (use any text editor).
As a more difficult example, let’s look at the interaction of the bacterial smallRNA RybB and its target mRNA ompN. First we’ll try predicting the binding site using RNAduplex:
$ RNAduplex < RybB.seq >RybB >ompN .((((..((((((.(((....((((((((..(((((.((..((.((....((((..(((((((((((..((((((& .))))))..))))))).)))).....))))....)).)).)).))).))..))))........))))..))).)))))).)))). 5,79 : 80,164 (-34.60)
Note, that the predicted structure spans almost the full length of the RybB small RNA. Compare the predicted interaction to the structures predicted for RybB and ompN alone, and ask yourself whether the predicted interaction is indeed plausible.
Below the structure of ompN on the left and RybB on the right side. The respective binding regions predicted by RNAduplex are marked in red.
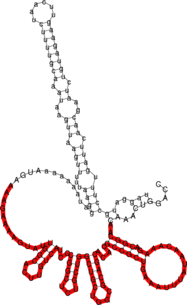

GCCAC-----TGCTTTTCTTTGATGTCCCCATTTT-GTGGA-------GC-CCATCAACCCCGCCATTTCGGTT---CAAG-GTTGGTGGGTTTTTT ||| |||| |||||| ||| ||||| |||| || ||| || || || |||| |||| || ||| |||||| -40.30 AGGTCAAACAACGGC-AGAAACAATATT--TAAAGTCGCCGCACACGACGCGGTCGTCGGT-CGTCTCGGCCCTACTGTTCACGGTTATGAAAAGAAACC-3'
Compare the RNAduplex prediction with the interaction predicted by RNAcofold, RNAup and the
handcrafted prediction you see above.
Consensus Structure Prediction
Sequence co-variations are a direct consequence of RNA base pairing rules and can be deduced to alignments. RNA helices normally contain only 6 out of the 16 possible combinations: the Watson-Crick pairs GC, CG, AU, UA, and the somewhat weaker wobble pairs GU and UG. Mutations in helical regions therefore have to be correlated. In particular we often find “compensatory mutations” where a mutation on one side of the helix is compensated by a second mutation on the other side, e.g. a CG pair changes into a UA pair. Mutations where only one pairing partner changes (such as CG to UG) are termed “consistent mutations”.
The Program RNAalifold
RNAalifold generalizes the folding algorithm for sequence alignments, treating
the entire alignment as a single “generalized sequence”. To assign an energy to a structure on such a
generalized sequence, the energy is simply averaged over all sequences in the alignment. This average
energy is augmented by a covariance term, that assigns a bonus or penalty to every possible base pair
based on the sequence
variation in columns
and of
the alignment.Compensatory mutations are a strong indication of structural conservation, while
consistent mutations provide a weaker signal. The covariance term used by RNAalifold
therefore assigns a bonus of 1 kcal/mol to each consistent and 2 kcal/mol for each
compensatory mutation. Sequences that cannot form a standard base pair incur a penalty of
kcal/mol. Thus, for every possible consensus pair between two columns
and
of the alignment a
covariance score
is computed by counting the fraction of sequence pairs exhibiting consistent and compensatory
mutations, as well as the fraction of sequences that are inconsistent with the pair. The weight of the
covariance term relative to the normal energy function, as well as the penalty for inconsistent
mutations can be changed via command line parameters.
Apart from the covariance term, the folding algorithm in RNAalifold is essentially the
same as for single sequence folding. In particular, folding an alignment containing just
one sequence will give the same result as single sequence folding using RNAfold. For
sequences of length
the required CPU
time scales as
while memory requirements grow as the square of the sequence length. Thus RNAalifold is in
general faster than folding each sequence individually. The main advantage, however, is that
the accuracy of consensus structure predictions is generally much higher than for single
sequence folding, where typically only between 40% and 70% of the base pairs are predicted
correctly.
Apart from prediction of MFE structures RNAalifold also implements an algorithm to compute the
partition function over all possible (consensus) structures and the thermodynamic equilibrium
probability for each possible pair. These base pairing probabilities are useful to see structural
alternatives, and to distinguish well defined regions, where the predicted structure is most likely
correct, from ambiguous regions.
As a first example we’ll produce a consensus structure prediction for the following four tRNA sequences.
$ cat four.seq
>M10740 Yeast-PHE GCGGAUUUAGCUCAGUUGGGAGAGCGCCAGACUGAAGAUUUGGAGGUCCUGUGUUCGAUCCACAGAAUUCGCA >K00349 Drosophila-PHE GCCGAAAUAGCUCAGUUGGGAGAGCGUUAGACUGAAGAUCUAAAGGUCCCCGGUUCAAUCCCGGGUUUCGGCA >K00283 Halobacterium volcanii Lys-tRNA-1 GGGCCGGUAGCUCAUUUAGGCAGAGCGUCUGACUCUUAAUCAGACGGUCGCGUGUUCGAAUCGCGUCCGGCCCA >AF346993 CAGAGUGUAGCUUAACACAAAGCACCCAACUUACACUUAGGAGAUUUCAACUUAACUUGACCGCUCUGA
RNAalifold uses aligned sequences as input. Thus, our first step will be to align the sequences. We
use clustalw2 in this example, since it’s one of the most widely used alignment programs and has
been shown to work well on structural RNAs. Other alignment programs can be used (including
programs that attempt to do structural alignment of RNAs), but the resulting multiple sequence
alignment must be in Clustal format. Get clustalw2 and install it as you have done it with the
other packages: http://www.clustal.org/clustal2
-
Prepare a sequence file (use file
four.seqand copy it to your working directory) - Align the sequences
- Compute the consensus structure from the alignment
-
Inspect the output files
alifold.out,alirna.ps,alidot.ps -
For comparison fold the sequences individually using
RNAfold
$ clustalw2 four.seq > four.out
Clustalw2 creates two more output files, four.aln and four.dnd. For RNAalifold you need the.aln file.
$ RNAalifold -p four.aln $ RNAfold -p < four.seq
RNAalifold output:
__GCCGAUGUAGCUCAGUUGGG_AGAGCGCCAGACUGAAAAUCAGAAGGUCCCGUGUUCAAUCCACGGAUCCGGCA__
..(((((((..((((.........)))).(((((.......))))).....(((((.......))))))))))))...
minimum free energy = -15.12 kcal/mol (-13.70 + -1.43)
..(((((({..((((.........)))).(((((.......))))).....(((((.......)))))}))))))...
free energy of ensemble = -15.75 kcal/mol
frequency of mfe structure in ensemble 0.361603
..(((((((..((((.........)))).(((((.......))))).....(((((.......))))))))))))... -15.20 {-13.70 + -1.50}
RNAfold output:
>M10740 Yeast-PHE
GCGGAUUUAGCUCAGUUGGGAGAGCGCCAGACUGAAGAUUUGGAGGUCCUGUGUUCGAUCCACAGAAUUCGCA
((((((((........((((.((((((..((((...........))))..))))))..))))..)))))))). (-21.60)
((((((({...,,.{,((((.((((((..((((...........))))..))))))..))))),)))))))). [-23.20]
((((((((.........(((.((((((..((((...........))))..))))))..)))...)))))))). {-20.00 d=9.63}
frequency of mfe structure in ensemble 0.0744065; ensemble diversity 15.35
>K00349 Drosophila-PHE
[...]The output contains a consensus sequence and the consensus structure in dot-bracket notation. The consensus structure has an energy of kcal/mol, which in turn consists of the average free energy of the structure kcal/mol and the covariance term kcal/mol. The strongly negative covariance term shows that there must be a fair number of consistent and compensatory mutations, but in contrast to the average free energy it’s not meaningful in the biophysical sense.
Compare the predicted consensus structure with the structures predicted for the individual
sequences using RNAfold. How often is the correct “clover-leaf” shape predicted?
For better visualization, a structure annotated alignment or color annotated structure drawing can
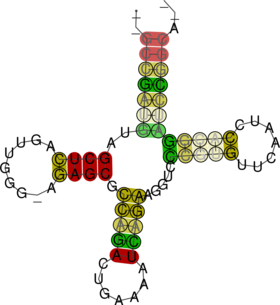
be generated by using the --aln and --color options of RNAalifold.
$ RNAalifold --color --aln four.aln $ gv aln.ps & $ gv alirna.ps &
RNAalifold Output Files4 sequence; length of alignment 78 alifold output 6 72 0 99.8% 0.007 GC:2 GU:1 AU:1 33 43 0 98.9% 0.033 GC:2 GU:1 AU:1 31 45 0 99.0% 0.030 CG:3 UA:1 15 25 0 98.9% 0.045 CG:3 UA:1 5 73 1 99.7% 0.008 CG:2 GC:1 13 27 0 99.1% 0.042 CG:4 14 26 0 99.1% 0.042 UA:4 4 74 1 99.5% 0.015 CG:3 [...]
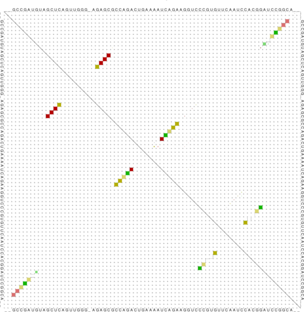
The last output file produced by RNAalifold -p, named alifold.out, is a plain text file with detailed
information on all plausible base pairs sorted by the likelihood of the pair. In the example above we see
that the pair
has no inconsistent sequences, is predicted almost with probability 1, and occurs as a GC pair in two
sequences, a GU pair in one, and a AU pair in another.
RNAalifold automatically produces a drawing of the consensus structure in Postscript format and
writes it to the file “alirna.ps”. In the structure graph consistent and compensatory mutations are
marked by a circle around the variable base(s), i.e. pairs where one pairing partner is encircled
exhibit consistent mutations, whereas pairs supported by compensatory mutations have both bases
marked. Pairs that cannot be formed by some of the sequences are shown gray instead of black. In
the example given, many pairs show such inconsistencies. This is because one of the sequences
(AF346993) is not aligned well by clustalw.
Note, that subsequent calls to RNAalifold will overwrite any existing output alirna.ps
(alidot.ps, alifold.out) files in the current directory. Be sure to rename any files you want to
keep.
The consensus structure computed by RNAalifold will contain only pairs that can be formed by
most of the sequences. The structures of the individual sequences will typically have additional base
pairs that are not part of the consensus structure. Moreover, ncRNA may exhibit a highly conserved
core structure while other regions are more variable. It may therefore be desirable to produce
structure predictions for one particular sequence, while still using covariance information from other
sequences.
This can be accomplished by first computing the consensus structure for all sequences using
RNAalifold, then folding individual sequences using RNAfold -C with the consensus structure as a
constraint. In constraint folding mode RNAfold -C allows only base pairs to form which are
compatible with the constraint structure. This resulting structure typically contains most of the
constraint (the consensus structure) plus some additional pairs that are specific for this
sequence.
The
refold.pl (find it in the Progs folder) script removes gaps and
maps the consensus structure to each individual sequence.
$ RNAalifold RNaseP.aln > RNaseP.alifold $ gv alirna.ps $ refold.pl RNaseP.aln RNaseP.alifold | head -3 > RNaseP.cfold $ RNAfold -C --noLP < RNaseP.cfold > RNaseP.refold $ gv E-coli_ss.ps
If you compare the refolded structure (E-coli_ss.ps) with the structure you get by simply folding the E.coli sequence in the RNaseP.seq file (RNAfold --noLP) you find a clear rearrangement.
In cases where constrained folding results in a structure that is very different from the consensus, or if the energy from constrained folding is much worse than from unconstrained folding, this may indicate that the sequence in question does not really share a common structure with the rest of the alignment or is misaligned. One should then either remove or re-align that sequence and recompute the consensus structure.
Note that since RNase P forms sizable pseudo-knots, a perfect prediction is impossible in this case.
Structural Alignments
Manually correcting Alignments
As the tRNA example above demonstrates, sequence alignments are often unsuitable as a basis for determining consensus structures. As a last resort, one may always try manually correcting an alignment. Sequence editors that are structure-aware may help in this task. In particular the SARSE http://sarse.kvl.dk/ editor, and theralee-mode for emacs
http://personalpages.manchester.ac.uk/staff/sam.griffiths-jones/software/ralee/ are
useful.
After downloading the ralee-files extract them and put them in a folder called ~/Tutorial/Progs/ralee.
Now read the 00README file and follow the instructions. If you don’t find an “.emacs”
file in your home directory execute the following command to copy it from the Data
directory.
$ cp Data/dot.emacs ~/
Next try correcting the ClustalW generated alignment (four.aln) from the example above. For this
we first have to convert it to the Stockholm format. Fortunately the formats are similar.
Make a copy of the file add the correct header line and the consensus structure from
RNAalifold:
$ cp four.aln four.stk $ emacs four.stk ..... $ cat four.stk
The final alignment should look like:
# STOCKHOLM 1.0 K00349 --GCCGAAAUAGCUCAGUUGGG-AGAGCGUUAGACUGAAGAUCUAAAGGUCCCCGGUUCAAUCCCGGGUUUCGGCA-- K00283 GGGCCG--GUAGCUCAUUUAGGCAGAGCGUCUGACUCUUAAUCAGACGGUCGCGUGUUCGAAUC--GCGUCCGGCCCA M10740 --GCGGAUUUAGCUCAGUUGGG-AGAGCGCCAGACUGAAGAUUUGGAGGUCCUGUGUUCGAUCCACAGAAUUCGCA-- AF346993 --CAGAGUGUAGCUUAAC---ACAAAGCACCCAACUUACACUUAGGAGAUUUCAACUUAACUUGACCGCUCUGA---- #=GC SS_cons ..(((((((..((((.........)))).(((((.......))))).....(((((.......))))))))))))... //
Now use the functions under the edit menu to improve the alignment, the coloring by structure should help to highlight misaligned positions.
Automatic structural alignments
Next, we’ll compute alignments using two structural alignment programs: LocARNA and T-Coffee.
LocARNA is an implementation of the Sankoff algorithm for simultaneous folding and alignment (i.e.
it will generate both alignment and consensus structure). T-Coffee uses a progressive alignment
algorithm.
Download LocARNA from http://www.bioinf.uni-freiburg.de/Software/LocARNA/, extract and
install it in your Progs folder and eventually add it to your path variable or copy it into the
corresponding directory.
Both programs can read the fasta file four.seq.
$ mlocarna --alifold-consensus-dp four.seq
[...]
M10740 GCGGAUUUAGCUCAGUUGGG-AGAGCGCCAGACUGAAGAUUUGGAGGUCCUGUGUUCGAUCCACAGAAUUCGCA
K00349 GCCGAAAUAGCUCAGUUGGG-AGAGCGUUAGACUGAAGAUCUAAAGGUCCCCGGUUCAAUCCCGGGUUUCGGCA
K00283 GGGCCGGUAGCUCAUUUAGGCAGAGCGUCUGACUCUUAAUCAGACGGUCGCGUGUUCGAAUCGCGUCCGGCCCA
AF346993 CAGAGUGUAGCUUAAC---ACAAAGCACCCAACUUACACUUAGGAGAUU-UCAACUUAA-CUUGACCGCUCUGA
alifold (((((((..((((.........)))).(((((.......))))).....(((((.......)))))))))))).
(-52.53 = -21.58 + -30.95)Get T-Coffee from the github page https://github.com/cbcrg/tcoffee. There is a detailed
information how you should download and install the software in the given README.md.
Go to the downloads directory and use the provided installer by typing
$ cd Tutorial/downloads $ git clone git@github.com:cbcrg/tcoffee.git tcoffee $ cd tcoffee/compile/ $ make t_coffee $ cp t_coffee ~/Tutorial/Progs/
Afterwards align the four.seq using t_coffee and compare the output with the one given by
LocARNA.
$ t_coffee four.seq > t_coffee.out
[t_coffee.out]
CLUSTAL FORMAT for T-COFFEE 20150925_14:18 [http://www.tcoffee.org] [MODE: ],
CPU=0.00 sec, SCORE=739, Nseq=4, Len=74
M10740 GCGGAUUUAGCUCAGUU-GGGAGAGCGCCAGACUGAAGAUUUGGAGGUCC
K00349 GCCGAAAUAGCUCAGUU-GGGAGAGCGUUAGACUGAAGAUCUAAAGGUCC
K00283 GGGCCGGUAGCUCAUUUAGGCAGAGCGUCUGACUCUUAAUCAGACGGUCG
AF346993 CAGAGUGUAGCUUAAC---ACAAAGCACCCAACUUACACUUAGGAGAUUU
***** * * *** *** * * *
M10740 UGUGUUCGAUCCACAGAAUUCGCA
K00349 CCGGUUCAAUCCCGGGUUUCGGCA
K00283 CGUGUUCGAAUCGCGUCCGGCCCA
AF346993 CAACUUAACUUGACCG--CUCUGA
** *Use RNAalifold to predict structures for all your alignments (ClustalW, handcrafted, T-Coffee, and LocARNA) and compare them. The handcrafted and LocARNA alignments should be essentially perfect.
Other interesting approaches to structural alignment include CMfinder, dynalign, and
stemloc.
Noncoding RNA gene prediction
Prediction of ncRNAs is still a challenging problem in bioinformatics. Unlike protein coding genes, ncRNAs do not have any statistically significant features in primary sequences that could be used for reliable prediction. A large class of ncRNAs, however, depend on a defined secondary structure for their function. As a consequence, evolutionarily conserved secondary structures can be used as characteristic signal to detect ncRNAs. All currently available programs for de novo prediction make use of this principle and are therefore, by construction, limited to structured RNAs.
-
QRNA(Eddy & Rivas, 2001) -
ddbRNA(di Bernardo, Down & Hubbard, 2003) -
MSARi(Coventry, Kleitman & Berger, 2004) -
AlifoldZ(Washietl & Hofacker, 2004) -
RNAz(Washietl, Hofacker & Stadler, 2005) -
EvoFold(Pedersen et al, 2006)
QRNA
QRNA analyzes pairwise alignments for characteristic patterns of evolution. An alignment is
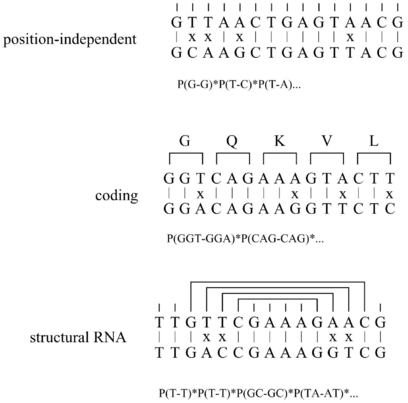
scored by three probabilistic models: (i) Position independent, (ii) coding, (iii) RNA. The
independent and the coding model is a pair hidden Markov model. The RNA model is a pair
stochastic context-free grammar. First, it calculates the prior probability that, given a
model, the alignment is observed. Second, it calculates the posterior probability that,
given an alignment, it has been generated by one of the three models. The posterior
probabilities are compared to the position independent background model and a “winner” is
found.
QRNA reads pairwise alignments in MFASTA format (i.e. FASTA format with gaps)
-
Use the files in
qrna-2.0.3d.tar.gzlocated in theData/programs-folder shipped with the tutorial -
don’t forget to set the
QRNADBenvironment variable
(e.g. export QRNADB=$HOME/Tutorial/Data/programs/qrna-2.0.3d/lib/) and add it to your.bashrc -
follow the instructions in the
INSTALLdocument and make the binaries -
create the directory
~/Tutorial/Progs/qrnaand move the binaries located in thesrc/sub-directory into this folder and add it to your.bashrc
(e.g. export PATH=${HOME}/Tutorial/Progs:${PATH}:${HOME}/Tutorial/Progs/qrna) -
first read the help text (option
-h). -
for advanced use of
QRNAread theuserguide.pdfshipped with the package (in thedocumentationfolder -
-atellsQRNAto print the alignment
$ eqrna -h $ eqrna -a Data/qrna/tRNA.fa $ eqrna -a Data/qrna/coding.fa
[...]
Divergence time (variable): 0.214132 0.208107 0.203995
[alignment ID = 72.37 MUT = 23.68 GAP = 3.95]
length alignment: 76 (id=72.37) (mut=23.68) (gap=3.95)
posX: 0-75 [0-72](73) -- (0.18 0.30 0.36 0.16)
posY: 0-75 [0-75](76) -- (0.14 0.34 0.37 0.14)
DA0780 GGGCTCGTAGCTCAGCT.GGAAGAGCGCGGCGTTTGCAACGCCGAGGCCT
DA0940 GGGCCGGTAGCTCAGCCTGGGAGAGCGTCGGCTTTGCAAGCCGAAGGCCC
DA0780 GGGGTTCAAATCCCCACGGGTCCA..
DA0940 CGGGTTCGAATCCCGGCCGGTCCACC
[..]AlifoldZ
AlifoldZ is based on an old hypothesis: functional RNAs are thermodynamically
more stable than expected by chance. This hypothesis can be statistically tested by calculating
-scores: Calculate the
MFE of the native
RNA and the mean
and standard deviation
of the background distribution of a large number of random (shuffled) RNAs. The normalized
-score
expresses how many standard deviations the native RNA is more stable than random
sequences.
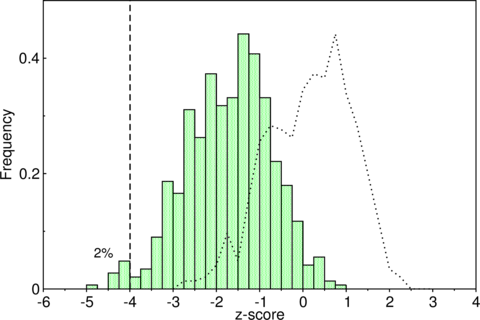
Unfortunately, most ncRNAs are not significantly more stable than the background. See for example the distribution of -scores of some tRNAs, where the overlap of real (green bars) and shuffled (dashed line) tRNAs is relatively high.
The overlap of real (bars) and shuffled (dashed line) tRNAs is relatively
high.
AlifoldZ calculates -scores
for consensus structures folded by RNAalifold. This significantly improves the
detection performance compared to single sequence folding.
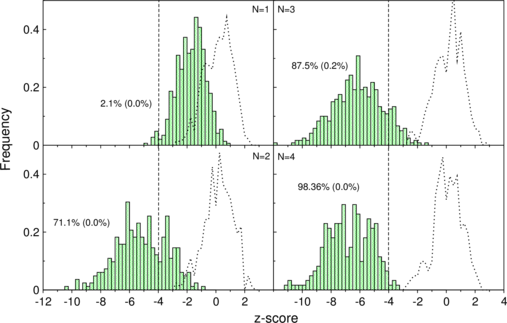
The separation of real and shuffled tRNAs gets evident with more sequences in the
alignment.
-
Use the tarball
alifoldz_adopted.tar.gzlocated in theData/programs/-folder shipped with the tutorial -
Copy the files into your
Progsdirectory (It’s just one single Perl script which needsRNAfoldandRNAalifoldand an important perl module located in the Math subdirectory)
$ cp -r alifoldz.pl Math/ ~/Tutorial/Progs/ -
add the perl module to your
PERL5LIBvariable in the.bashrc
$ export PERL5LIB=$HOME/Tutorial/Progs/:$PERL5LIB - test the tool
$ alifoldz.pl -h $ alifoldz.pl < Data/alifoldz/miRNA.aln $ alifoldz.pl -w 120 -x 100 < Data/alifoldz/unknown.aln
RNAz
* New version by Someone who loves RNAz. This part of the tutorial is based on the RNAz 1.0 version which is obsolete quite a while already!!! *AlifoldZ has some shortcomings that limits its usefulness in practice: The
-scores are
not deterministic, i.e. you get a different score each time you run AlifoldZ. To get stable
-scores
you need to sample a large number of random alignments which is computationally
expensive. Moreover, AlifoldZ is extremely sensitive to alignment errors.
The program RNAz overcomes these problems by using a different approach
to asses a multiple sequence alignment for significant RNA structures. It
is based on two key innovations: (i) The structure conservation index
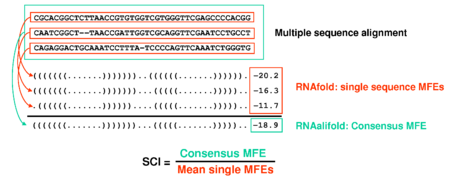
(SCI) to measure structural conservation in an alignment and (ii)
-scores
that are calculated by regression without sampling. Both measures are combined to
an overall score that is used to classify an alignment as “structured RNA” or
“other”.
-
The structure conservation index is an easy way to normalize an
RNAalifoldconsensus MFE.
- The mean and standard deviation of random samples of a given sequence are functions of the length and the base composition:
- It is therefore be possible to calculate -scores by solving this 5 dimensional regression problem.
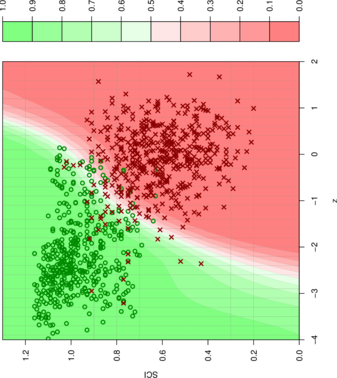
- A support vector machine learning algorithm is used to classify an alignment based on -score and structure conservation index.
Installation is done according to the instructions used by the ViennaRNA Package.
Just use the --prefix option as mentioned earlier and add the PATH to .bashrc
- RNAz is available at: http://www.tbi.univie.ac.at/~wash/RNAz
-
Package includes the core program
RNAzin ISOC, a set of helper programs in Perl, and an extensive manual.
* where to get examples from (RNAz install package) - commands work with v2 but txt needs to be adopted *
-
RNAzreads one or more multiple sequence alignments inclustalwor MAF format.
$ RNAz --help $ RNAz tRNA.aln $ RNAz --both-strands --predict-strand tRNA.maf
-
RNAzis limited to a maximum alignment length of 400 columns and a maximum number of 6 sequences. To process larger alignments a set of Perl helper scripts are used. - Selecting one or more subsets of sequences from an alignment with more than 6 sequences:
$ rnazSelectSeqs.pl miRNA.maf |RNAz $ rnazSelectSeqs.pl --num-seqs=4 --num-samples=3 miRNA.maf |RNAz
- Scoring long alignments in overlapping windows:
$ rnazWindow.pl --window=120 --slide=40 unknown.aln \
| RNAz --both-strandsLarge scale screens
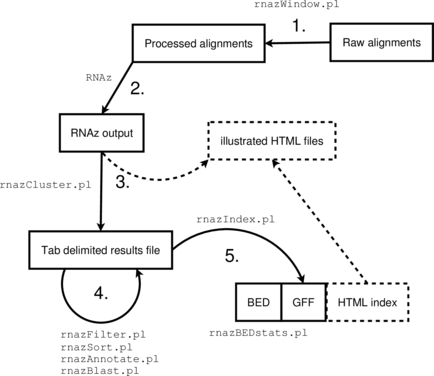
The RNAz package provides a set of Perl scripts that implement a complete analysis
pipeline suitable for medium to large scale screens of genomic data.
- Obtain or create multiple sequence alignments in MAF format
-
Run through the
RNAzpipeline:
-
Align Epstein Barr Virus genome (Acc.no: NC_007605) to two related
primate viruses (Acc.nos: NC_004367, NC_006146) using
multizand run it through theRNAzpipeline. * where are this data comeing from? file from NCBI differs from those hidden in genefinding/rnaz/herpes * -
Analyze snoRNA cluster in the human genome for conserved RNA
structures: download pre-computed alignments from the UCSC genome
browser and run it through the
RNAzpipeline
-
multizandblastzare available here: http://www.bx.psu.edu/miller_lab/ -
Download the viral genomes in FASTA format and reformat the header
strictly according to the rules given in the
multizdocumentation (http://www.bx.psu.edu/miller_lab/dist/tba_howto.pdf), e.g.: -
You have to edit the ”multiz ”
Makefileand replaceCFLAGS = -Wall -Wextra -Werror
with
CFLAGS = -Wall -Wextra #-Werror
and then simply use the
makecommand to compile both programs.
* dont understand what to do *
>NC_007605:genome:1:+:149696 AGAATTCGTCTTGCTCTATTCACCCTTACTTTTCTTCTTGCCCGTTCTCTTTCTTAGTAT GAATCCAGTATGCCTGCCTGTAATTGTTGCGCCCTACCTCTTTTGGCTGGCGGCTATTGC CGCCTCGTGTTTCACGGCCTCAGTTAGTACCGTTGTGACCGCCACCGGCTTGGCCCTCTC ACTTCTACTCTTGGCAGCAGTGGCCAGCTCATATGCCGCTGCACAAAGGAAACTGCTGAC ACCGGTGACAGTGCTTACTGCGGTTGTCACTTGTGAGTACACACGCACCATTTACAATGC ATGATGTTCGTGAGATTGATCTGTCTCTAACAGTTCACTTCCTCTGCTTTTCTCCTCAGT CTTTGCAATTTGCCTAACATGGAGGATTGAGGACCCACCTTTTAATTCTCTTCTGTTTGC [...]
-
To get a multiple alignment a phylogenetic tree and the following three steps
are necessary:
-
Run
blastzeach vs. each - Combine blastz results to multiple sequence alignments
- Project raw alignments to a reference sequence.
-
Run
- The corresponding commands:
all_bz - "((NC_007605 NC_006146) NC_004367)" | bash tba "((NC_007605 NC_006146) NC_004367)" \ *.sing.maf raw-tba.maf maf_project raw-tba.maf NC_007605 > final.maf
- Note: The tree is given in NEWICK like format with blanks instead of commas. The sequence data files must be named exactly like the names in this tree and in the FASTA headers.
- First the alignments are filtered and sliced in overlapping windows:
$ rnazWindow.pl < final.maf > windows.maf
-
RNAzis run on these windows:
$ RNAz --both-strands --show-gaps --cutoff=0.5 windows.maf \
> rnaz.out- Overlapping hits are combined to “loci” and visualized on a web-site:
$ rnazCluster.pl --html rnaz.out > results.dat
- The predicted hits are compared with available annotation of the genome:
$ rnazAnnotate.pl --bed annotation.bed results.dat \
> results_annotated.dat- The results file is formatted in a HTML overview page:
$ rnazIndex.pl --html results_annotated.dat \
> results/index.html-
rnazIndex.plcan be used to generate a BED formatted annotation file which can be analyzed usingrnazBEDstats.pl(after sorting, for the case the input alignments were unsorted)”
$ rnazIndex.pl --bed results.dat | \
rnazBEDsort.pl | rnazBEDstats.pl- RNAzfilter.pl can be used to filter the results by different criteria. In this case it gives us all loci with P0.9”:
$ rnazFilter.pl "P>0.9" results.dat | \
rnazIndex.pl --bed | \
rnazBEDsort.pl | rnazBEDstats.pl- To get an estimate on the (statistical) false positives one can repeat the complete screen with randomized alignments:
$ rnazRandomizeAln final.maf > random.maf
- Go to the UCSC genome browser (http://genome.ucsc.edu) and go to “Tables”. Download “multiz17” alignments in MAF format for the region: chr11:93103000-93108000
- The Perl scripts are run in the same order as in Example 1:
$ rnazWindow.pl --min-seqs=4 region.maf > windows.maf
$ RNAz --both-strands --show-gaps --cutoff=0.5 windows.maf \
> rnaz.out
$ rnazCluster.pl --html rnaz.out > results.dat
$ rnazAnnotate.pl --bed annotation.bed results.dat \
> results_annotated.dat
$ rnazIndex.pl --html results_annotated.dat \
> results/index.html- The results can be exported as UCSC BED file which can be displayed in the genome browser:
$ rnazIndex.pl --bed --ucsc results.dat > prediction.bed
