

Next: Phylogenetic Analysis
Up: 300632 UE Exercises for
Previous: Pairwise Sequence Alignment
The goal of this section is to compare and evaluate the robustness of
several multiple sequence alignment programs. As test-dataset
Rieske iron sulfur protein sequences from a number of species are
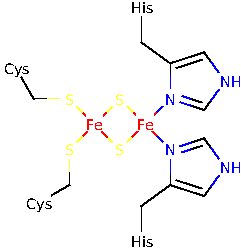
used. The critical functional site of this protein family bind an
iron-sulfur
![$[\text{Fe}_2\text{S}_2]$](img4.png) cluster with the two known consensus
amino acid binding motifs CXHXGC and CXXH. Investigate,
in particular, the ability of the multiple sequence alignment programs to
discover the two conserved binding motifs as well as to correctly align the
rest of the protein sequences.
cluster with the two known consensus
amino acid binding motifs CXHXGC and CXXH. Investigate,
in particular, the ability of the multiple sequence alignment programs to
discover the two conserved binding motifs as well as to correctly align the
rest of the protein sequences.

- Download the protein sequences with the following accession numbers
in FASTA format from NCBI Entrez database:
P08067, P20788, AAD55565, P08980, P23136, AAC84018, AAF02198.
- Use the progressive alignment program ClustalW
http://www.ch.embnet.org/software/ClustalW.html to align the
sequences. (Save resulting multiple alignment in
ClustalW format).
- Check the quality of the alignment visually and by using the
alignment quality evaluation web service CORE
http://igs-server.cnrs-mrs.fr/Tcoffee/tcoffee_cgi/index.cgi.
- Use the profile-based alignment program MultiAlign
http://bioinfo.genotoul.fr/multalin/ to align the
sequences. (Note: conversions between different file formats can be
done by the program Readseq
http://iubio.bio.indiana.edu/cgi-bin/readseq.cgi. Use
CORE to check the quality of the multiple alignment).
- Use the semi-exhaustive program DCA
http://bibiserv.techfak.uni-bielefeld.de/dca/submission.html to
align the sequences. (Do a quality evaluation as above).
- Use the iterative tree-based program PRRN
http://www.genome.ist.i.kyoto-u.ac.jp/~aln_user/cgi-bin/prrn.pl
to align the sequences. (Do a quality evaluation as above).
- Finally use the program T-Coffee
http://igs-server.cnrs-mrs.fr/Tcoffee/tcoffee_cgi/index.cgi to
align the sequences. (Do a quality evaluation as above).
- Carefully compare the results from the different methods. Can you
identify the most reasonable alignment? Which method appears to be the
best?
Next: Phylogenetic Analysis
Up: 300632 UE Exercises for
Previous: Pairwise Sequence Alignment
Christoph Flamm
2009-01-06