A map of structural RNAs in the Human genome
Introduction
This page shows the results of a large scale comparative screen for structural RNAs in the human genome as described in the manuscript
Stefan Washietl, Ivo L. Hofacker, Melanie Lukasser, Alexander Hüttenhofer and Peter F. Stadler.
Nature Biotechnology,23:1383-90.
You can download and read the paper and the supplementary material. The screen is based on the program RNAz.
Annotation data
The results can be downloaded as annotation tracks in BED format compatible with the UCSC genome browser. The coordinates refer to the "hg17" assembly (May 2004). We provide six tracks corresponding to different phylogenetic subsets and significance levels (See manuscript for details). The corresponding sequences can be downloaded in FASTA format. Important note: Since our predictions at this stage have no strand assignment the FASTA files contain always the forward strand sequence. If you use these sequences for further analysis you should also consider the reverse complement.
- Set 1: Conserved at least in human/mouse/rat/dog
- P>0.5: set1_50.bed | set1_50.fa.gz
- P>0.9: set1_90.bed | set1_90.fa.gz
- Set 2: Conserved at least in human/mouse/rat/dog and chicken
- P>0.5: set2_50.bed | set2_50.fa.gz
- P>0.9: set2_90.bed | set2_90.fa.gz
- Set 3: Conserved at least in human/mouse/rat/dog/chicken and fugu or zebrafish
- P>0.5: set3_50.bed | set3_50.fa.gz
- P>0.9: set3_90.bed | set3_90.fa.gz
You can upload the tracks here, or you can directly open the genome browser with Set-1 (P>90) by clicking this link.
Browsing the database of predicted structures
We scanned long alignments in sliding windows. Overlapping hits were combined into clusters. Each annotated feature in the track corresponds to a cluster of RNAz hits.
The annotation track is linked to a detailed description for each cluster (example). It provides an overview of the predicted structures in the cluster, the results of a database search of all major ncRNA databases, and annotation relative to known/predicted protein coding genes and ESTs.
In addition, there is a detailed page for each detected structure in the cluster (example). It presents the input alignment, the RNAz results and a consensus secondary structure model.
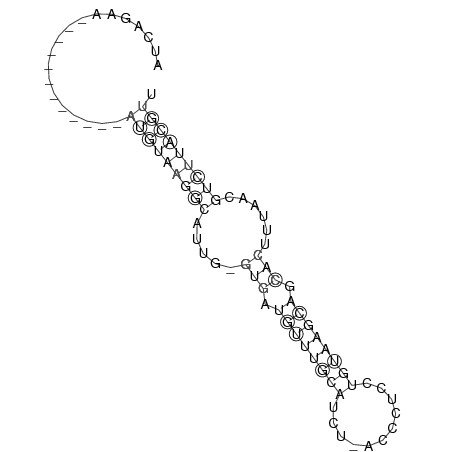
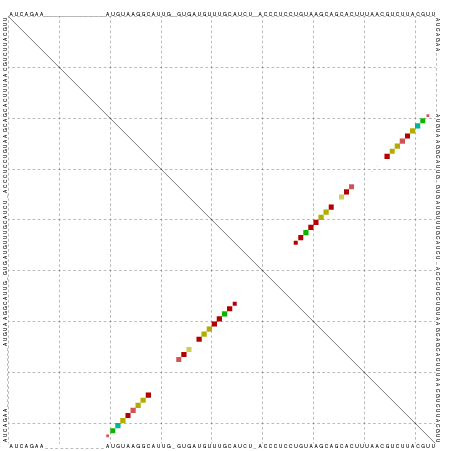
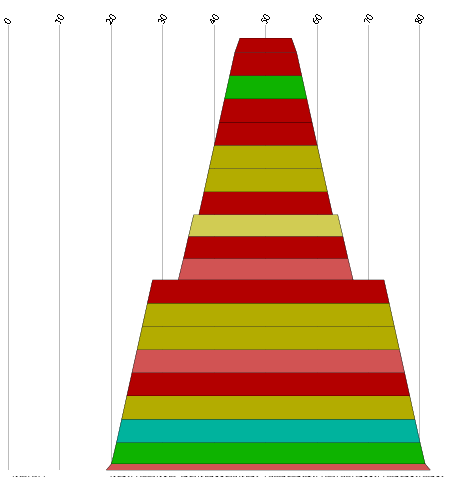
The consensus secondary structure is computed by RNAalifold, the program which RNAz is based on. It computes the most likely structure and base pair probabilities for each base pairs. The results are shown as secondary structure graph (example) and, alternatively, as "dot-plot" (example) or Hogeweg mountain plot (example).
{kind=link}
{kind=link}
{kind=link}
In the secondary structure graph, variable positions are marked with circles (one circle: consistent mutation, two circles: compensatory mutation). In addition, the number of alternative base combinations/predicted pair are color-coded. Incompatible pairs in the consensus structure are indicated by pale colors.
| Types of pairs | |||||||
| 1 | 2 | 3 | 4 | 5 | 6 | ||
| Incompatible pairs | 0 | ||||||
| 1 | |||||||
| 2 | |||||||
Examples
- snRNAs: U11 and U12, all other snRNAs are not in our input set because they were masked by RepeatMasker
- RNAaseP, a good example where the structure is highly conserved (the SCI of 0.69 is very high given the pairwise identity of only 69.37%), but not exceptionally stable (z-score only -0.83).
- In the case of many H/ACA snoRNAs and miRNAs, both conservation and stabilility scores are high.
- Most of the predicted structures are not supported by a large number of consistent/compensatory mutations as seen in the ideal examples above. The concept of the SCI, however, does not only rely on consistent/compensatory mutations. Also the mutation pattern outside stems is implicitly taken into account. Together with the stability score RNAz can thus detect also subtle signals (C/D snoRNA).
Selected subsets
Our screen has a significant false positive rate (we estimate appr. 20% for the P>0.9 cut-off). However, high probable candidates can be filtered by applying more stringent criteria. For example, we selected hits with mean-pairwise <85%, SCI>0.7, z<-3. This gives us 1221 candidates which are exceptionally stable and structurally conserved. The list presents the top 100 ot this set sorted by the number of compensatory mutations supporting the consensus structure:
One can also filter the hits by secondary structure motifs. As described in the manuscript, we can detect a number H/ACA snoRNA candidates which have resisted computational and experimental detection so far:
We compared our hits with a recent transcriptional map based on tiling array technology (Cheng et al.,Science 308:1149-1154, 2005). Here is a list of 50 selected hits we found in intergenic regions (more than 10kb from any known gene based on "known" Gene track from UCSC and without plausible protein gene prediction) that coincide with hits from the array data. We used the cumulative map of PolyA+ and PolyA- transcripts from 8 different cell lines.
We plan to post more subsets/subscreens here in the future.